Впервые ребенка с трисомией одной из хромосом группы D описали в 1960 г. Patau, Smith, Therman, Inhorn и Wagner. Это была девочка, родившаяся у здоровых 25-летних родителей. Мать на VI месяце беременности перенесла гриппоподобное заболевание. Девочка родилась с цианозом. Впервые ее увидел один из исследователей (Smith) через месяц после рождения (рис. 131).
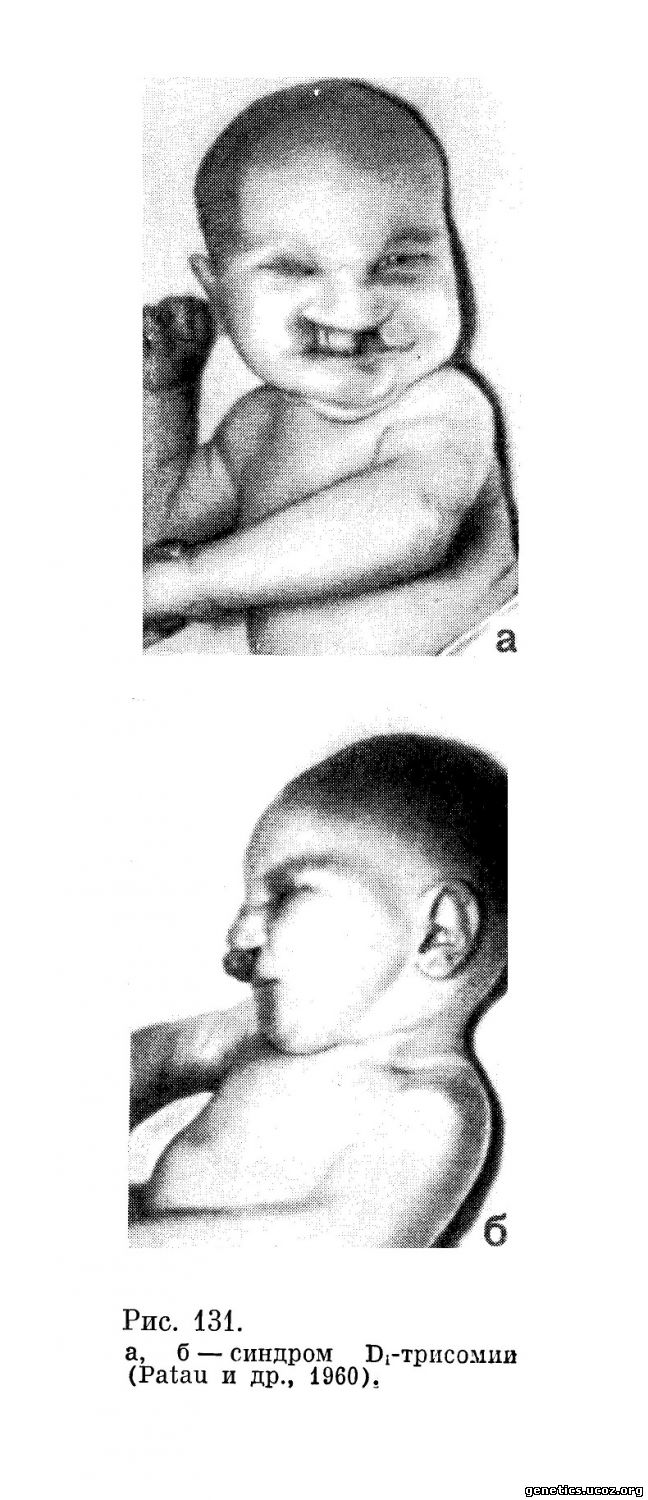
Девочка была хорошо упитана, но у нее имелись множественные аномалии, среди которых особенно обращали на себя внимание анофтальмия, незаращение верхней губы и неба, наличие лишнего пальца на левой стопе. Внешний вид черепа был нормальный, однако рентгенологическое исследование выявило плохое окостенение черепных костей и наличие больших промежутков между ними. В глазницах пальпировалась неорганизованная ткань глазного яблока. Незаращение губы было двусторонним,, больше выражено справа. Незаращение неба было полным. Клиническое исследование показало наличие врожденного порока сердца. На коже отмечались многочисленные гемангиомы. Девочка была глуха. У исследователей возникло предположение, что здесь, возможно, имеется атипичный синдром Дауна. Однако исследование дерматоглифики показало резкое отличие от того, что обычно бывает при синдроме Дауна. При цитогенетическом исследовании клеток костного мозга было обнаружено 47 хромосом, причем лишней оказалась хромосома из группы D (13—15) 47, XX, D+ (рис. 132).

Вскоре были опубликованы описания еще нескольких случаев сочетания множественных врожденных пороков развития с трисомией одной из хромосом группы D. Постоянство цитогенетических находок и особое сочетание уродств подтвердили точку зрения первых исследователей, что во всех случаях речь идет о новом синдроме, связанном с трисомией одной из хромосом группы D (13—15). После открытия Lejeune, показавшего связь синдрома Дауна с трисомией одной из хромосом группы G, это был второй синдром, обусловленный хромосомной патологией. Однако Smith (1964) в своем обзоре указал, что первые сообщения, судя до описанию аномалий с иллюстрациями, были сделаны еще в 1657 г, анатомом и патологом Bartholin, в 1892 г. — Kundrat, а в 1959 г. несколько случаев описал Jakovleff. Несомненно, многие патологоанатомы наблюдали случаи, которые называют синдромом Di-трисомии, или синдромом Патау. Во всяком случае Сопеп и Erkman (19666) обнаружили в Канаде несколько таких случаев в протоколах патологоанатомического архива.
К 1963 г. было описано около 15 случаев этого заболевания, а к 1966 г.— уже около 40.
а) Цитогенетика
Хромосомы группы D относятся к большим акроцентрическим хромосомам. Они все являются спутничными и вступают в ассоциации как между собой, так и с малыми акроцентриками группы G. Короткие плечи этих хромосом состоят из гетерохроматина. Хромосомы группы D не имеют, как правило, вторичных перетяжек. Радиоавтографические исследования показали, что хромосомы группы D, вовлеченные в трисомию, являются поздно репродуцирующимися (Junis, Hook, 1965). Точная идентификация хромосом в пределах этой группы невозможна.
б) Частота заболевания
Со времени первой публикации разными авторами приводились весьма различные данные о частоте синдрома Di-трисомии среди живорожденных. Это связано как с малым объемом выборки, обычно доступной исследователю, так и с несовершенством обработки материала. В Канаде, в штате Онтарио, Сопеп и Erkman (1966) провели обширное исследование на большой выборке живорожденных в достаточно стабильной популяции, демографически хорошо документированной и с показателями витальной статистики. Они располагали данными относительно 224 460 младенцев, данными патологоанатомических служб и демографической статистики штата Онтарио. Эти исследователи предварительно выяснили, что врачи достаточно точно могут регистрировать случаи синдрома Патау, во всяком случае диагнозы врачей довольно часто подтверждались цитогенетическими исследованиями. Однако, по мнению Сопеп и Erkman, около 5—25% случаев синдрома D-трисомии остаются нераспознанными. На основании этих данных, анализа литературы и собственных исследований, в ходе которых было выявлено 9 больных с синдромом Di-трисомии, авторы сделали вывод, что частота этого заболевания составляет 6,8 Х 10-5, или 1 случай на 14 500 живорождений. Работа Сопеп и Erkman является наиболее обстоятельной и специально посвящена вопросу о частоте заболевания, вследствие чего данные этих авторов можно считать наиболее обоснованными.
в) Возраст родителей
С того времени, как было обнаружено, что пожилой возраст матери коррелирует с увеличением частоты нерасхождения хромосомы 21, при обнаружении новых синдромов, связанных с трисомией, особое внимание обращается на возраст родителей. Хотя случаев синдрома Di-трисомии значительно меньше, чем случаев болезни Дауна, и поэтому нельзя сделать точных выводов о связи возраста матери с нерасхождением по одной из D-хромосом, все таки в литературе складывается мнение, что возраст матерей, родивших детей с синдромом Di-трисомии, относится к пожилому с акушерской точки зрения. Так, в уже цитировавшемся исследовании Сопеп и Erkman средний возраст матерей в случае Di-трисомии равен 32,8 года. Следует отметить, что средний возраст матерей в случае транслокационной формы синдрома Di-трисомии (D/D транслокация) — 24 года. Такое же явление отмечено и при транслокационных формах синдрома Еі-трисомии. Иными словами, в случае Di-трисомии, по-видимому, имеет место та же закономерность, что и в случае болезни Дауна: частота нерасхождения в мейозе увеличивается с возрастом, в то время как транслокационная форма чаще встречается у молодых матерей.
г) Наследственность
Никаких особенностей при генеалогическом исследовании в случаях Di- трисомии не обнаружено. При описании первого случая Patau и др. сообщили, что среди 224 родственников пробанда, распределенных по 3 поколениям, только у троих были отмечены аномалии разного рода, но не такие, которые наблюдались у пробанда. Иначе обстоит дело в случаях транслокационной формы этого синдрома. Клинически эти формы не различаются, однако при транслокационной форме возможно носительства транслокационной хромосомы и передача ее по наследству. В специальной работе Hecht, Braynt, Gruber и Townes (1964), изучившие 60 семей, из которых в 16 был пробанд с Di-трисомией, показали, что в семьях, где имеются больные с синдромом Дауна, чаще встречаются и больные с синдромом Di-трисомии. Они сообщили о семье, где был больной с синдромом Di-трисомии и с трисомией хромосомы 21 (болезнь Дауна). У матери больного отмечалась ненормальная дерматоглифика, у отца была типичная «обезьянья складка» и высоко расположенный трирадиус. У отца больного из 200 клеток в 7 была обнаружена трисомия по хромосоме 21. Эти факты свидетельствуют о том, что может существовать семейная тенденция к нерасхождению хромосом, связанная с неизвестными еще причинами.
д) Связь синдрома Di-трисомии с заболеваниями матери и действием внешних факторов
Ни в одном из описанных случаев не отмечена связь с каким-либо заболеванием матери или действием внешних факторов (облучение ионизирующей радиацией, контакт с вредными химическими агентами).
е) Клинические особенности синдрома
Беременность у матерей, родивших детей с синдромом Di-трисомии, протекает, как правило, нормально. Роды происходят в срок и без особых осложнений. Несмотря на нормальные роды, дети часто рождаются в асфиксии; как правило, отмечается цианоз. Аномалий плаценты не отмечается.
Внешний вид больных весьма характерен. Как правило, у них при рождении нормальные вес и размеры. Отмечается более или менее выраженная микроцефалия. Обращают на себя внимание низко расположенные (на уровне углов рта) и неправильно сформированные уши. Характерной аномалией является патология глаз. Чаще всего бывает микрофтальмия и анофтальмия, причем у одного индивидуума может быть с одной стороны микрофтальмия, с другой — анофтальмия. Часто отмечают колобому, размеры которой могут варьировать довольно значительно. Кроме колобомы, иногда бывает помутнение роговицы. Постоянным и обязательным признаком является незаращение губы (одностороннее или двустороннее) и полное отсутствие переднего мозга, выражающееся в арринэнцефалии (Laurence, 1964). Неоднократно описывалась также своеобразная аномалия боцефалия, которая может считаться достаточно типичной для этого синдрома (Buhler и др., 1962; Demeyer, 1964; Conen и др., 1966). Перечисленные аномалии Conen и Erkman обозначают признаками группы А и считают облигатными.
Опираясь на эти признаки, диагноз Di-трисомии можно поставить с большой степенью вероятности. К менее постоянным и труднее выявляемым признакам относятся полидактилия (нередко в виде кожного ридумента или обнаруживаемая только при рентгенологическом исследовании), аномалии почек, порок сердца.
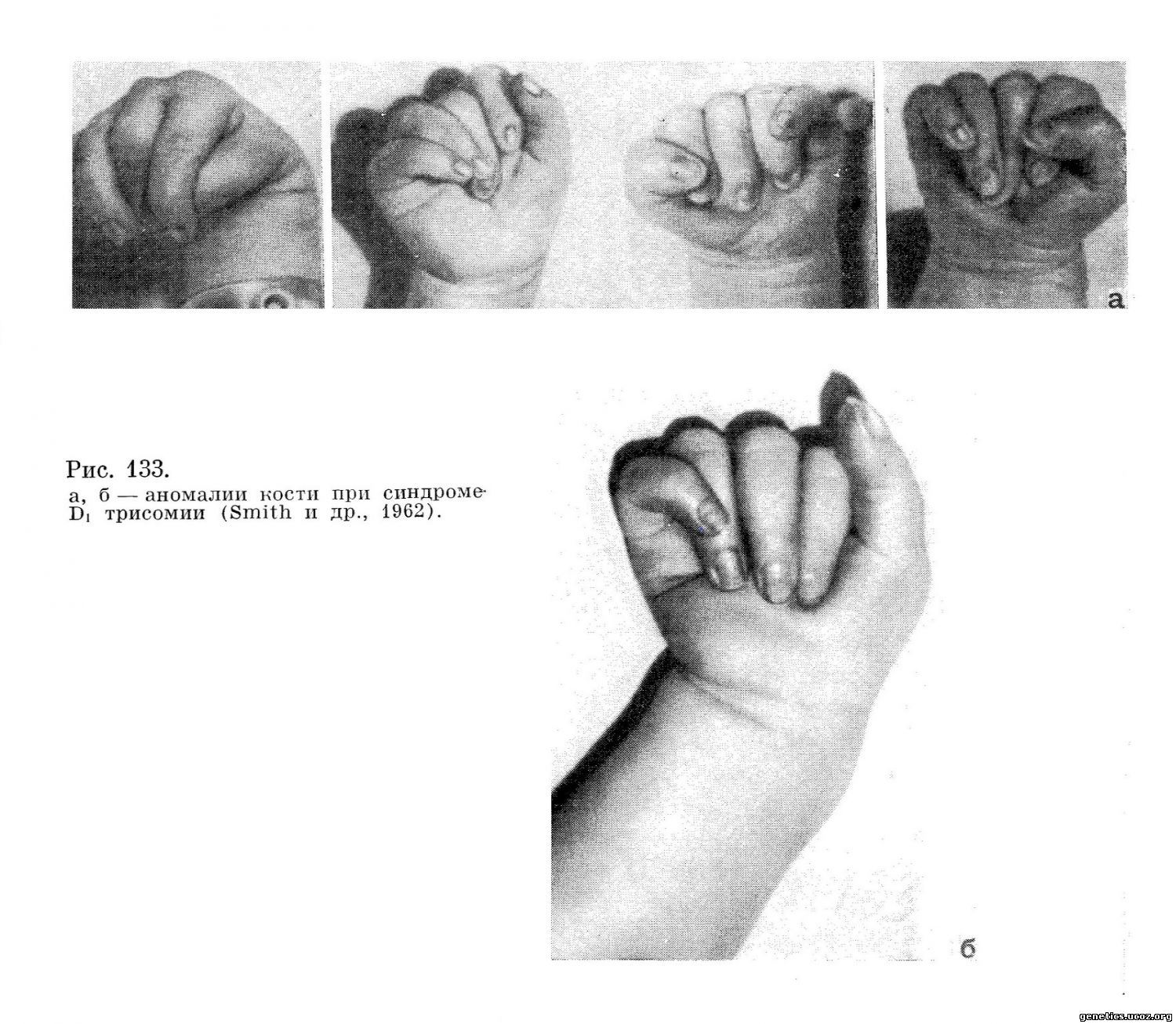
Дети с синдромом Di-трисомии обладают нормальным сосательным рефлексом. У них нет также значительных неврологических нарушений. В ряде случаев при рождении отмечается ульнарная флексия кисти и своеобразная аддукция больного пальца и мизинца (рис. 133). Иногда наблюдаются эпилептиформные припадки. Больные резко отстают в психомоторном развитии.
Продолжительность жизни детей невелика. По данным Conen и Erkman, большинство доживает до 100 дней. Причины смерти бывают самые разнообразные. Чаще всего дети умирают от случайных заболеваний. В ряде случаев причина смерти явно связана с анатомическими аномалиями (врожденный порок сердца).
ж) Патологическая анатомия
В ряде работ весьма подробно описана патологоанатомическая картина заболевания (Blank и др., 1964; Conen и др., 1962; Sergovich и др., 1963; Werburg, Mikkelsen, 1962; Rosenfield и др., 1962; Lee и др., 1966; Teller, Pfeiffer, 1964; Marin-Padilla и др., 1964).
Сердце. Часто находят дефекты межжелудочковой и межпредсердной перегородок. Все отделы сердца увеличены, имеется расширение всех камер. Так, в случае, описанном Blank и др., при весе тела больного 4015 г сердце весило 31 г после фиксации. Часто отмечается декстрапозиция сердца (Lee и др., 1966).
Легкие. Как правило, сформированы нормально, однако в них находят изменения, связанные с пороками сердца, — фиброз, умеренный ателектаз, явления хронической неспецифической пневмонии. При микроскопии в альвеолах обнаруживают экссудат с макрофагами и гранулоцитами.
Почки и мочеполовая система. В почках как правило, но не всегда обнаруживаются кистозные образования, чаще в корковом веществе. При микроскопическом исследовании находят микроцисты и в мозговом веществе.
Иногда (Blank и др., 1964) кисты окружены зоной интерстициального неспецифического воспаления с гранулоцитарной инфильтрацией. Наружные половые органы не изменены. Mirouzec с сотрудниками (1964) обнаружил при аномалии D хромосом синдром тестикулярной феминизации, a Therman и др. (1961) описали двух сестер с дисгенезией гонад при сочетании трисомии D и ХО.
Поджелудочная железа. Иногда (Blank и др., 1964) находят гетеротопию ткани селезенки в поджелудочную железу.
Пищеварительный тракт. Часто находят меккелев дивертикул.
Нервная система. Отмечается недоразвитие мозга; особенно характерно недоразвитие переднего мозга. Lee и др., (1966) описали больного с полным отсутствием обонятельных луковиц и тракта, с гипоплазированным зрительным трактом. У этого же больного они нашли дезорганизацию мозжечка, причем дезорганизованные массы мозжечковой ткани были гетеротопированы: клетки Пуркине были обнаружены в ядрах и на дне IV желудочка.
з) Лабораторные исследования
Кровь. Значительных изменений в клеточном составе и химических компонентах крови не обнаружено. Некоторые исследователи (Huełms и др., 1964; Stockenius и др., 1966) указывают, что в сегментоядерных лейкоцитах отмечаются выросты нуклеоплазмы наподобие «барабанных палочек». На этом основании указанные авторы предложили использовать исследование лейкоцитов для предварительной диагностики синдрома Di-трисомии. Однако другие исследователи (Mehes, 1966) полагают, что эти выросты не являются специфическими. Во всяком случае, этот тест не нашел пока практического применения. Гораздо более важными представляются биохимические аномалии, обнаруживаемые при синдроме Di-трисомии. Huehns и др., (1964), а также Powars с соавторами (1964) установили, что при синдроме Di-трисомии имеются аномалии гемоглобина, а именно персистенция фетального гемоглобина, уменьшение содержания гемоглобина А и А2. Такого же рода наблюдения принадлежат Stockenius с сотрудниками (1966), Lee и др. (1966). Последние авторы, кроме гемоглобина, изучали также содержание карбангидразы и эмбрионального і-антигена в эритроцитах. Обнаружено, что при этом заболевании количество карбангидразы резко снижено и имеется персистенция і-антигена.
Моча. Изменения в моче отмечаются лишь в случае развития пиелонефрита, что бывает довольно часто в связи с аномалиями почек.
Рентгенологическое исследование. Со стороны органов грудной клетки находят увеличение размеров сердца и усиление легочного рисунка. Рентгенологическое исследование конечностей помогает выявить слабо выраженную полидактилию.
и) Диагноз, прогноз и медико-генетическая консультация
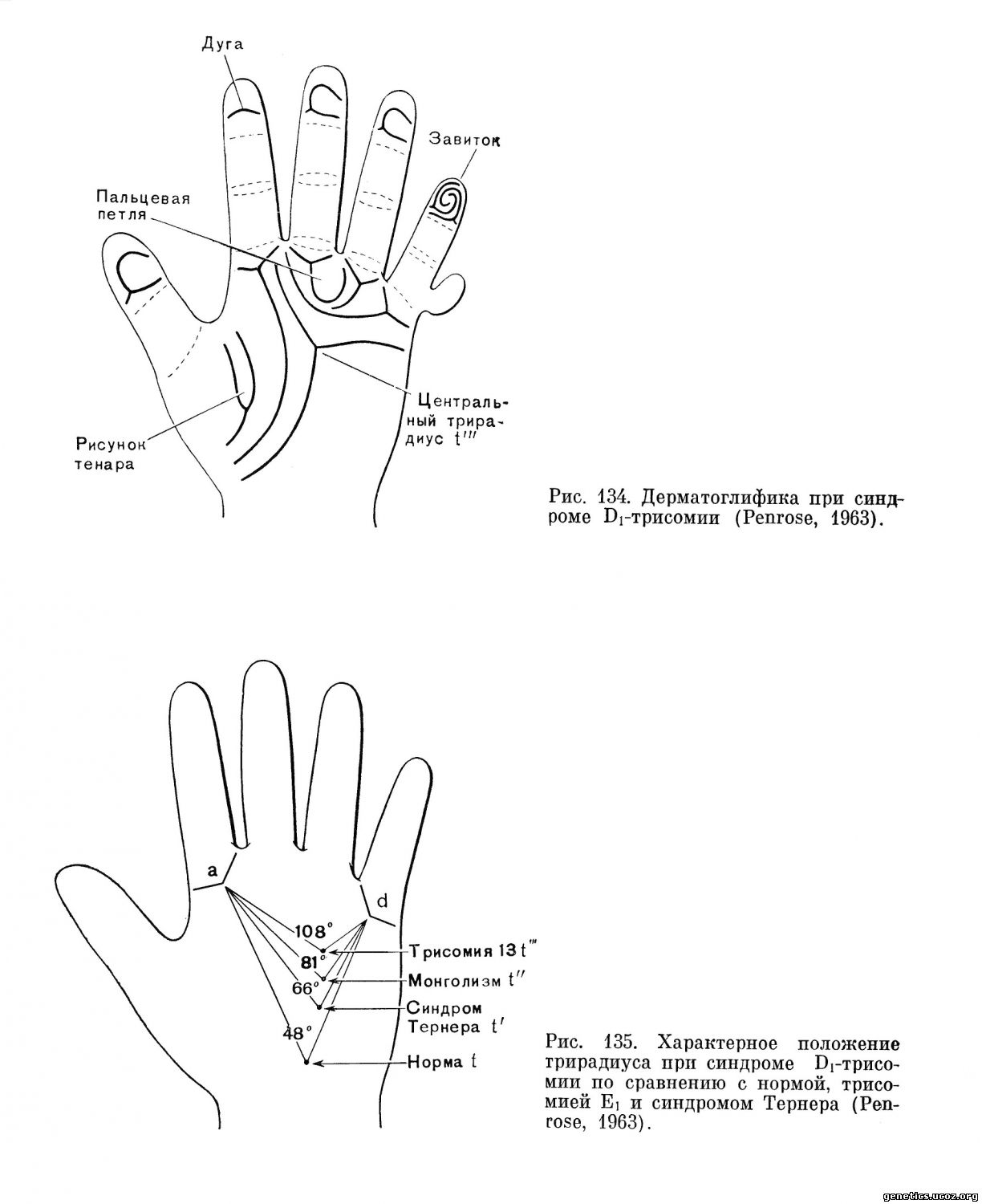
Диагноз заболевания основывается на клиническом исследовании больного. Наличие основных, облигатных симптомов делает диагноз высоковероятным. Дерматоглифическое исследование (Penrose, 1963; Uchida и др.,1962, Francois и др., 1966) еще более уточняет диагноз. Для дерматоглифики синдрома Di-трисомии характерны крайне дистальное расположение осевого трирадиуса (угол, образуемый осевым трирадиусом в вершине и трирадиусами оснований указательного пальца и мизинца, равен примерно 108° вместо 48° в норме и 81° при синдроме Дауна) (рис. 134, 135) поперечная ладонная складка; наличие папиллярных структур на возвышении большого пальца. Цитогенетическое исследование необходимо во всех случаях для верификации клинических данных и для исключения транслокаций и мозаицизма. Дифференциальный диагноз необходимо проводить с атипичными случаями болезни Дауна, с синдромом Еі-трисомии, а также с пороками развития, вызванными токсоплазмозом, краснухой и т. д.
В типичных случаях продолжительность жизни невелика — не больше года. Методов лечения не существует.
Ввиду того что транслокационная форма синдрома Di-трисомии практически неотличима по клиническим признакам от регулярной трисомии, обязательным является цитогенетическое исследование ребенка. В тех случаях, когда ребенок умер до этого, необходимо производить исследование хромосом у родителей. При транслокационной форме вероятность рождения ребенка с синдромом Di-трисомии достаточно высока — до 25%, в то время как при регулярной трисомии она, по-видимому, не выше, чем при болезни Дауна.
Однако точные цифры эмпирического риска ввиду малой частоты синдрома пока еще отсутствуют, но тем не менее следует полагать, что с увеличением возраста матери эта вероятность возрастает.
к) Нетипичные случаи
Имеются сообщения о мозаицизме, а также о случаях, когда аномалия D-хромосом обнаруживалась у здоровых людей. Описаны также случаи явного синдрома Di-трисомии, но с нормальным кариотипом.
Beęak с соавторами (1963) наблюдал семью, в которой два ребенка в возрасте 3 и 2 лет страдали врожденной генерализованной анальгезией. Их родители являлись двоюродными родственниками. При исследовании хромосом у детей было обнаружено, что часть клеток содержит нормальный набор хромосом, в то время как другие клетки были с трисомией D-хромосомы. Stone с соавторами (1966) описал 14-летнюю умственно отсталую девочку без внешних аномалий, с нормальной дерматоглификой, у которой 15% клеток содержали 47 хромосом и лишней была хромосома из группы 13—15. В связи с этим авторы полагают, что среди умственно отсталых детей может быть много мозаиков. Chandra и Hungerford (1963) описали аномальную D-хромосому у нормального мужчины и его дочери. В то же время Marshall с соавторами (1964) наблюдал больного с явными признаками синдрома Di-трисомии, но с нормальным кариотипом. Подобные случаи, хотя и являются редкими, представляют определенный интерес и о них необходимо помнить. Здесь, по-видимому, сталкиваются либо с трудно выявляемым мозаицизмом, либо с фенокопиями.
Отклонения от типичной картины синдрома могут быть и в случае сочетания трисомии D с другими хромосомными нарушениями (Therman и др., 1961).