Полисомия по половым хромосомам у лиц с мужским фенотипом касается избытка по сравнению с нормой числа Х-или Y-хромосом, а в некоторых случаях как Х-, так и Y-хромосом.
Клиническим проявлением полисомии по половым хромосомам у фенотипических мужчин является синдром Клайнфельтера. С цитогенетической точки зрения этот синдром можно разделить на три варианта:
1) полисомия по Х-хромосоме при моносомии по Y-хромосоме — 47, XXY; 48,. XXXY; 49, XXXXY;
2) полисомия по Y-хромосоме при моносомии по Х-хромосоме 47, XYY; 48, XYYY,
3) полисомия как по хромосомам Х так и Y - 48, XXYY; 49, XXXYY.
Вариант синдрома Клайнфельтера, полисомный по Х-хромосоме
Несмотря на множество цитогенетических вариантов, клиническая картина (фенотип) этих индивидуумов, имеющая некоторые особенности при различных генотипах, в то же время отражает общие черты, свойственные синдрому, описанному в 1942 г. Klinefelter с соавторами.
Синдром Клайнфельтера наблюдается у лиц с мужским фенотипом. Характерной особенностью синдрома является недоразвитие яичек. Неполноценность основной в отношении формирования пола эндокринной железы накладывает свой отпечаток на клиническое проявление болезни. Недостаточная гормональная активность яичек, обычно наблюдающаяся в период полового созревания, и возникающий в результате этого гормональный дисбаланс ведут к ненормальному для мужчин половому и физическому развитию и появлению ряда черт, свойственных женскому организму.
Больные с синдромом Клайнфельтера (рис. 102 и 103) обычно высокого роста, пропорции тела евнухоидные, отмечается тенденция к женскому строению скелета (узкие плечи, широкий таз), отложение жира по женскому типу, склонность к ожирению, иногда гинекомастия (одно- или двусторонняя), скудное оволосение в подмышечных впадинах, недостаточное оволосение на лобке с распределением роста волос по женскому типу. Растительность на лице очень скудная или совсем отсутствует.
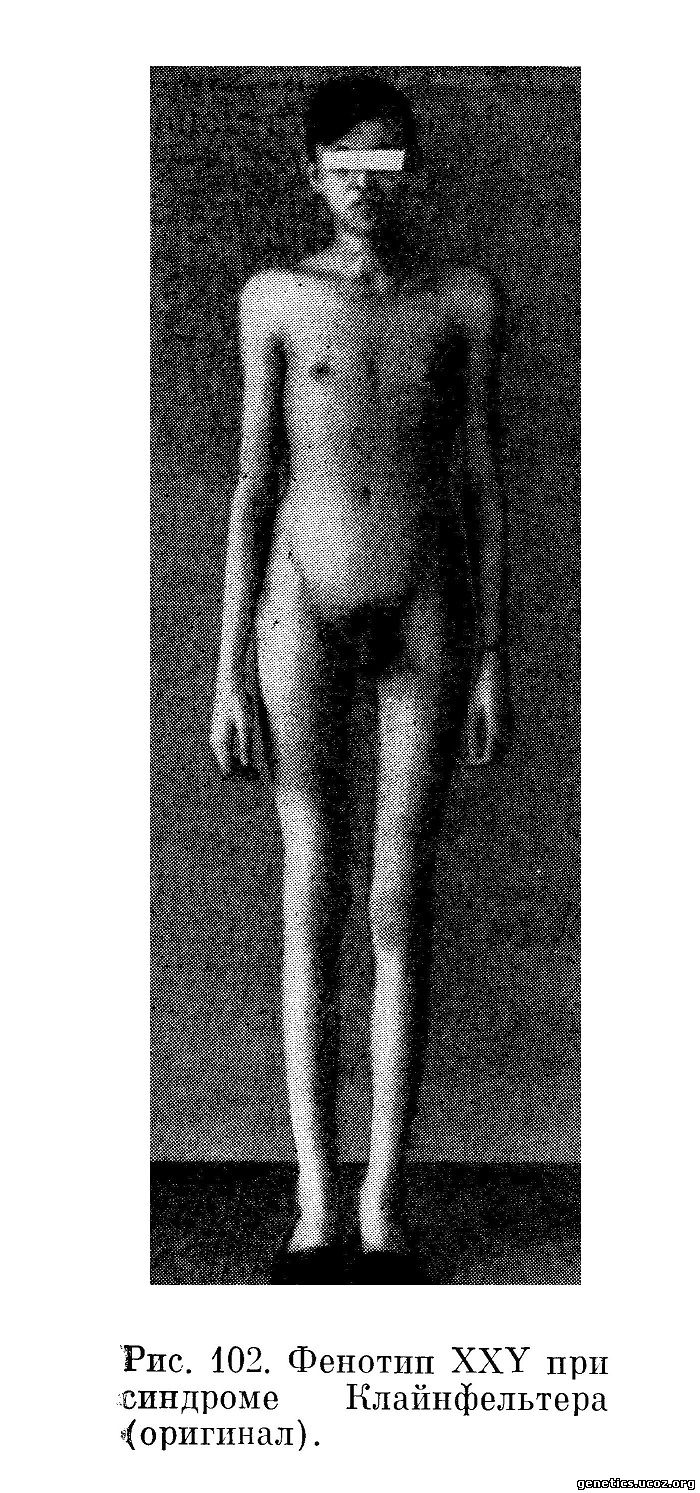
Половой член нормальных размеров или несколько уменьшен. Яички располагаются в мошонке, значительно уменьшены в размере (1—1,5 см), мягкие, при пальпации безболезненные. Простата уменьшена. В эякуляте обычно сперматозоидов обнаружить не удается. Гистология яичек обычно характеризуется различной степенью гиалиноза семенных канальцев, дегенерацией канальцевого эпителия, отсутствием или резким недоразвитием генеративных элементов, хотя в редких случаях находят зрелые формы сперматозоидов. В литературе описаны только единичные наблюдения над больными с синдромом Клайнфельтера, имевшими детей (Warburg, 1963). Отмечается обилие клеток Лейдига, однако это увеличение, вероятно, относительное, учитывая значительно меньший по сравнению с нормой размер яичек у этих больных. Выделение 17-кетостероидов с мочой находится на нижней границе нормы для мужчин.
Начиная с периода полового созревания, отмечается значительное повышение титра гонадотропинов в моче.
Синдром Клайнфельтера редко диагностируется до периода полового созревания ввиду отсутствия характерных симптомов, выявляющихся только к этому времени. Гистологическая картина яичек у этих больных до наступления полового созревания мало отличается от таковой у мальчиков того же возраста. Не исключено поэтому, что патологический сдвиг наступает в переходный период, и у индивидуумов, избежавших его по каким-либо причинам, может иметь место сперматогенез и нормальное потомство.
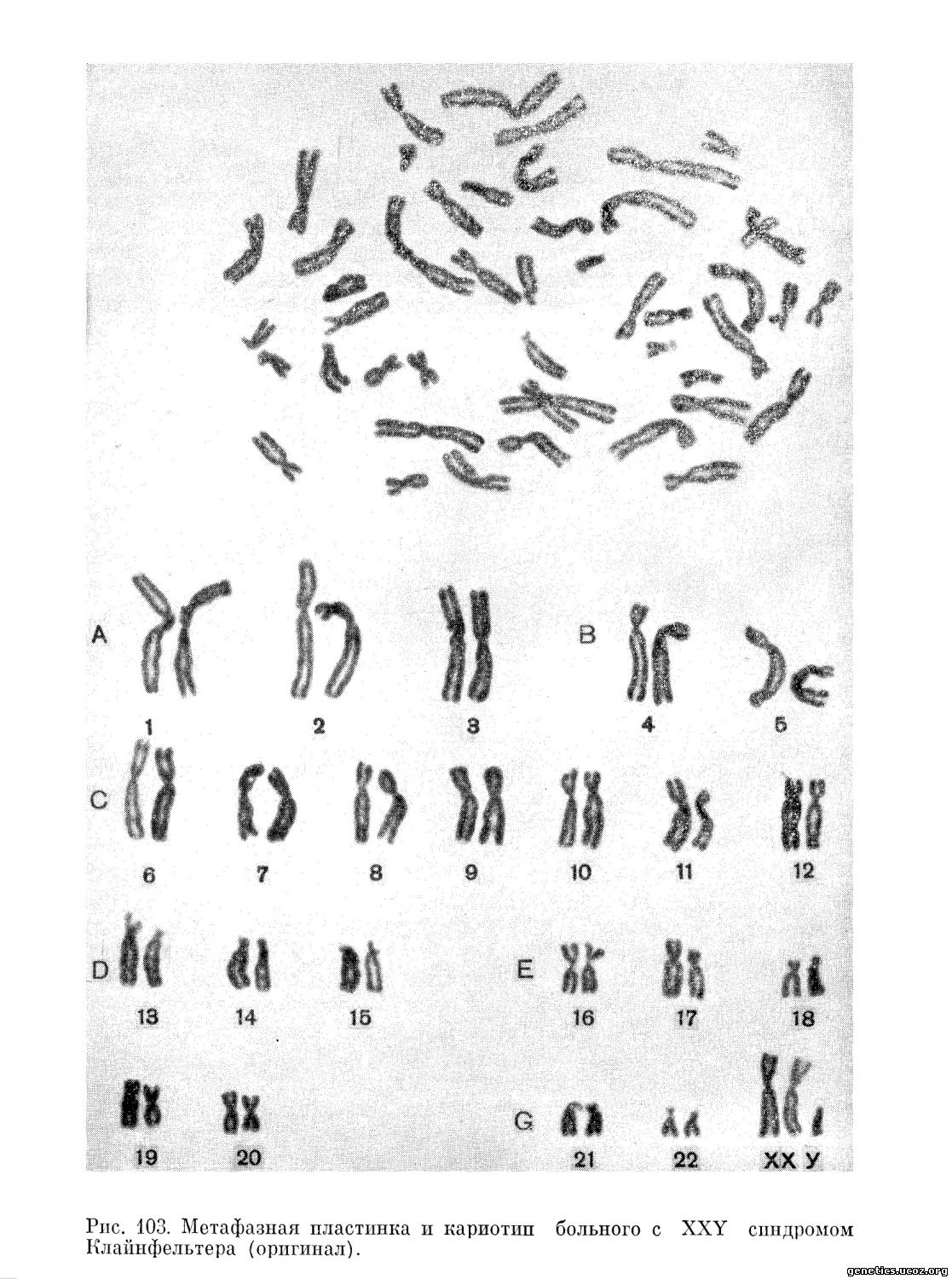
В 1956 г. Plunkett и Ватт (1956) показали, что у большинства мужчин с синдромом Клайнфельтера ядра слизистой оболочки полости рта содержат тельца полового хроматина. На этом основании было сделано заключение, что они имеют две Х-хромосомы являясь 46, XX или 47, XXY индивидуумами. Подтверждением этого предположения послужила низкая частота цветовой слепоты у этих пациентов по сравнению с нормальными мужчинами. Polani и др.. Strong, а также Ford с сотрудниками (1959) показали, что хроматинположительные пациенты с синдромом Клайнфельтера имеют XXY конституцию по половым хромосомам. В дальнейшем эти данные были подтверждены многочисленными исследователями (Ford и др., 1959; Bergman и др., 1960; Barr, Carr, 1960).
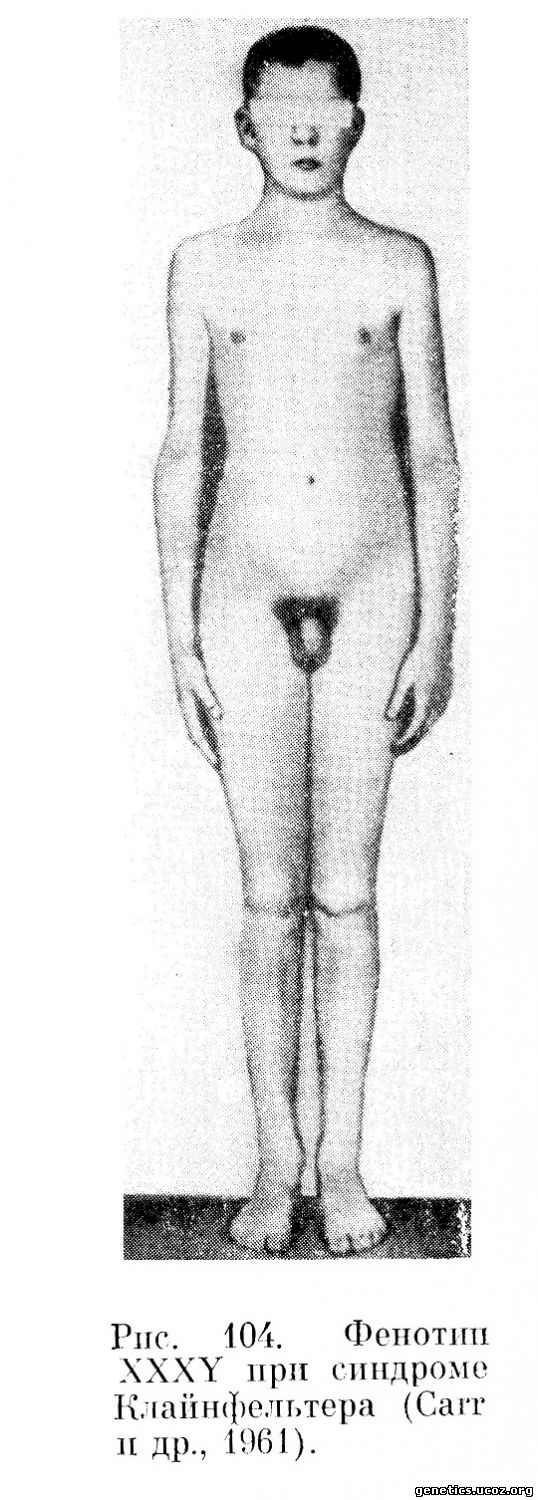
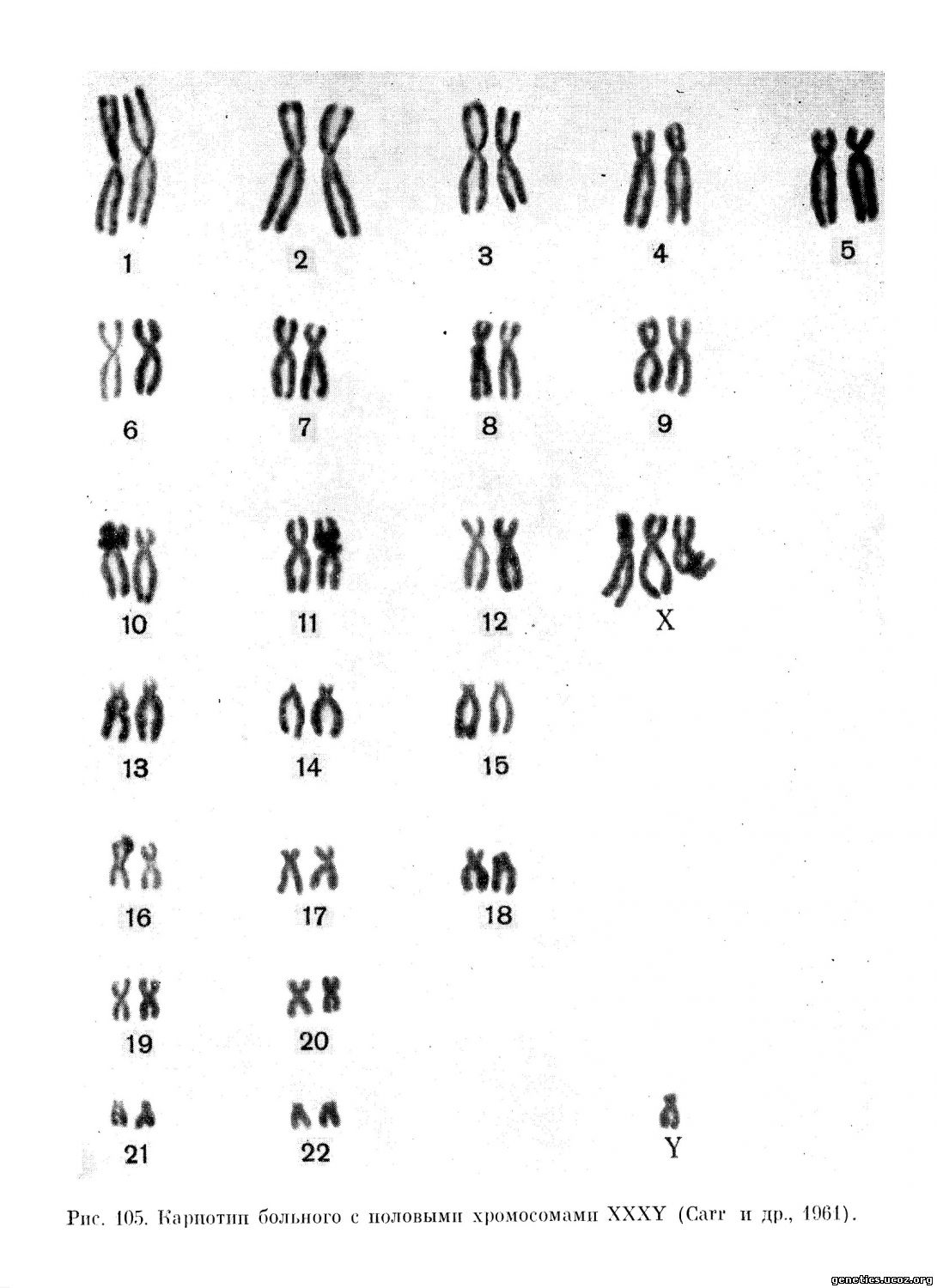
Вариант синдрома с двумя тельцами полового хроматина в соматических клетках и 48, XXXY конституцией половых хромосом описан Carr с сотрудниками (1961), Ferguson-Smith с сотрудниками (1960) и многими другими авторами. Клинически эти индивидуумы почти не отличались от 47, XXY, но имели значительную степень умственного недоразвития; (рис. 104 и 105).
49, XXXXY индивидуума (рис. 106) описали в 1960 г. Fraccaro и Lindsten (1960), а в дальнейшем ряд других исследователей (Miller и др., 1961; Turpin и др., 1962а). Для таких больных характерны тяжелая степень умственной отсталости и резкое недоразвитие яичек. Яички очень маленькие, часто неопустившиеся; у молодых мужчин гистологически находят скудное количество семенных трубочек и немного дегенеративных форм сперматозоидов или полное их отсутствие. У взрослых пациентов отмечается потеря тубулярной структуры, резко выраженный фиброз и полное отсутствие герминативных элементов. Часто встречаются аномалии развития костной системы (изменения коленных и локтевых суставов, радиоульнарный синостоз), отмечается увеличение нижней челюсти.
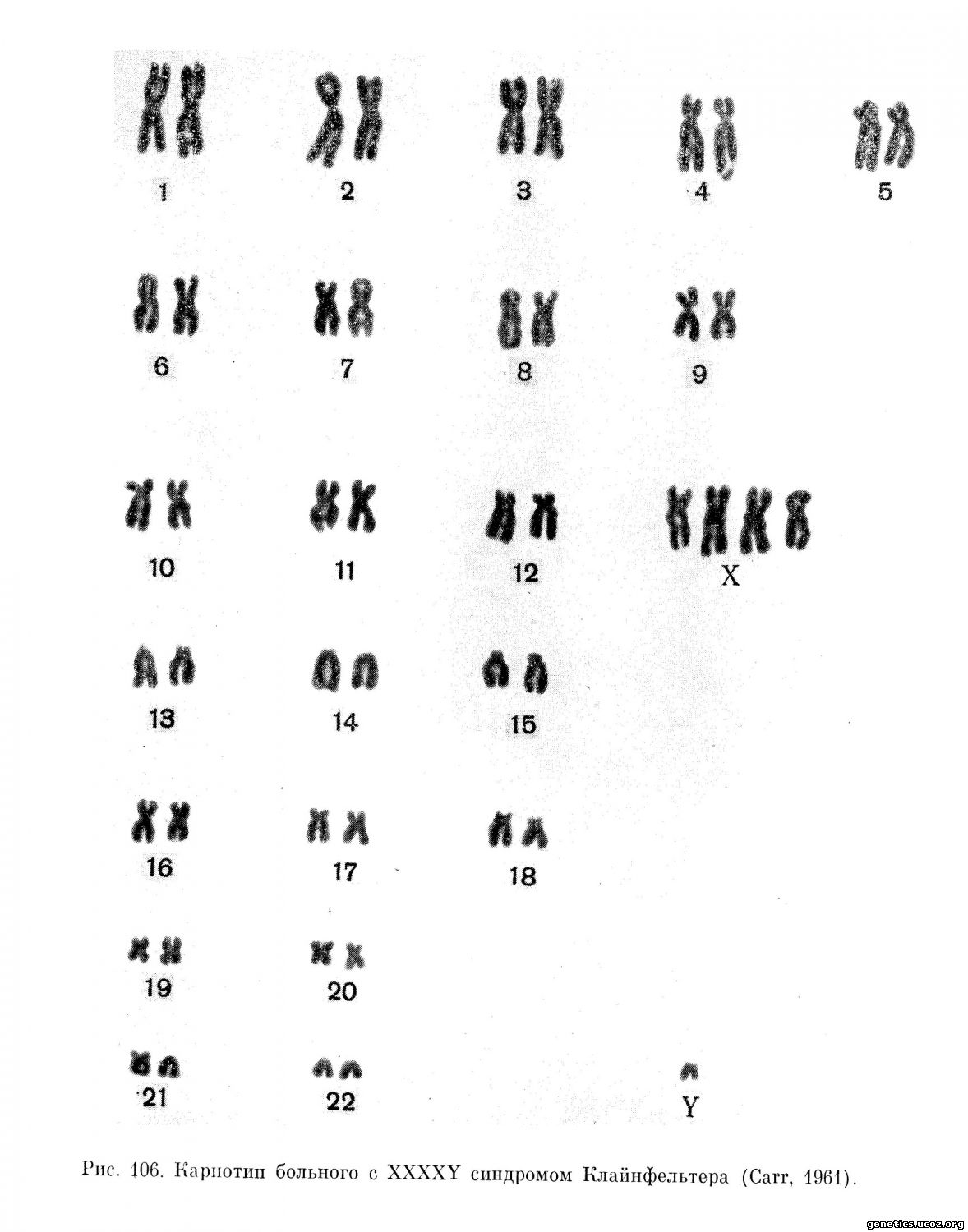
Как видно, в этой группе больных с увеличением числа Х-хромосом нарастают умственная отсталость, дегенеративные изменения в яичках, выявляются аномалии развития скелета.
Вариант синдрома Клайнфельтера, полисомный по Y-хромосоме
Наличие только одной Х-хромосомы и таким образом отсутствие полового хроматина, а также отсутствие в интерфазном ядре каких-либо косвенных признаков, указывающих на наличие более чем одной Y-хромосомы, не позволяет использовать экспресс-метод для диагностики этих пациентов. Фенотипические проявления у Индивидуумов, полисомных по Y-хромосоме при моносомии по Х-хромосоме, настолько полиморфны, что не позволяют выявить специфические клинические признаки у этой группы индивидуумов, объединенных общностью кариотипа. Описания отдельных случаев касаются индивидуумов различных возрастов, выявленных случайно, причем многие из сообщений не отличаются детальностью описания клинических особенностей. Дальнейшее накопление клинических наблюдений, вероятно, позволит более полно охарактеризовать фенотипические особенности этого варианта синдрома Клайнфельтера.
Hauschka с соавторами (1962) впервые описали XYY конституцию по половым хромосомам у фенотипически нормального 44-летнего мужчины рослом 183 см (рис. 107, 108). Отклонений от нормы в отношении его умственного развития отмечено не было. У данного субъекта не имелось никаких аномалий и он был выявлен случайно, при обследовании его неполноценных детей. В двух браках было 10 беременностей, из которых две закончились спонтанными абортами, пять—рождением нормальных мальчиков, одна — нормальной девочки, одна — рождением хроматинпозитивной девочки с первичной аменореей и отсутствием внутренних половых органов, у одной из дочерей была болезнь Дауна (трисомия по хромосоме 21); одна из беременностей закончилась рождением разнояйцевых близнецов, из которых один здоров, а другой умер в «синей асфиксии».
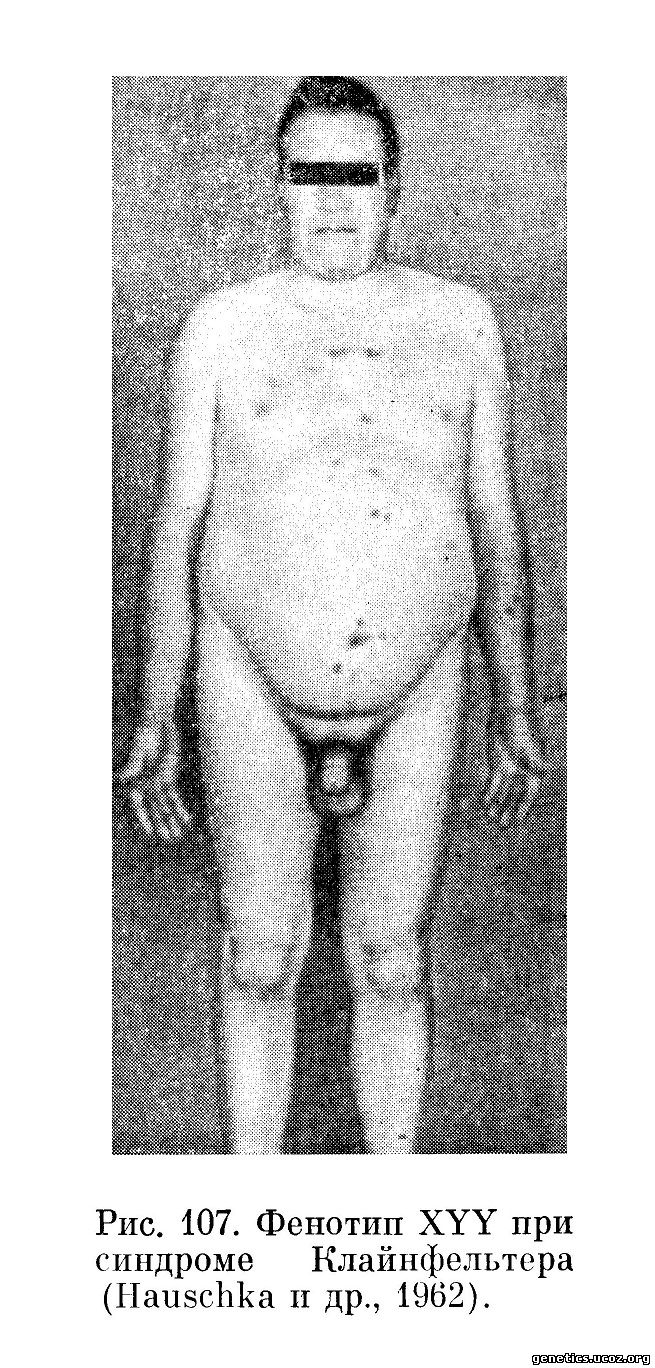
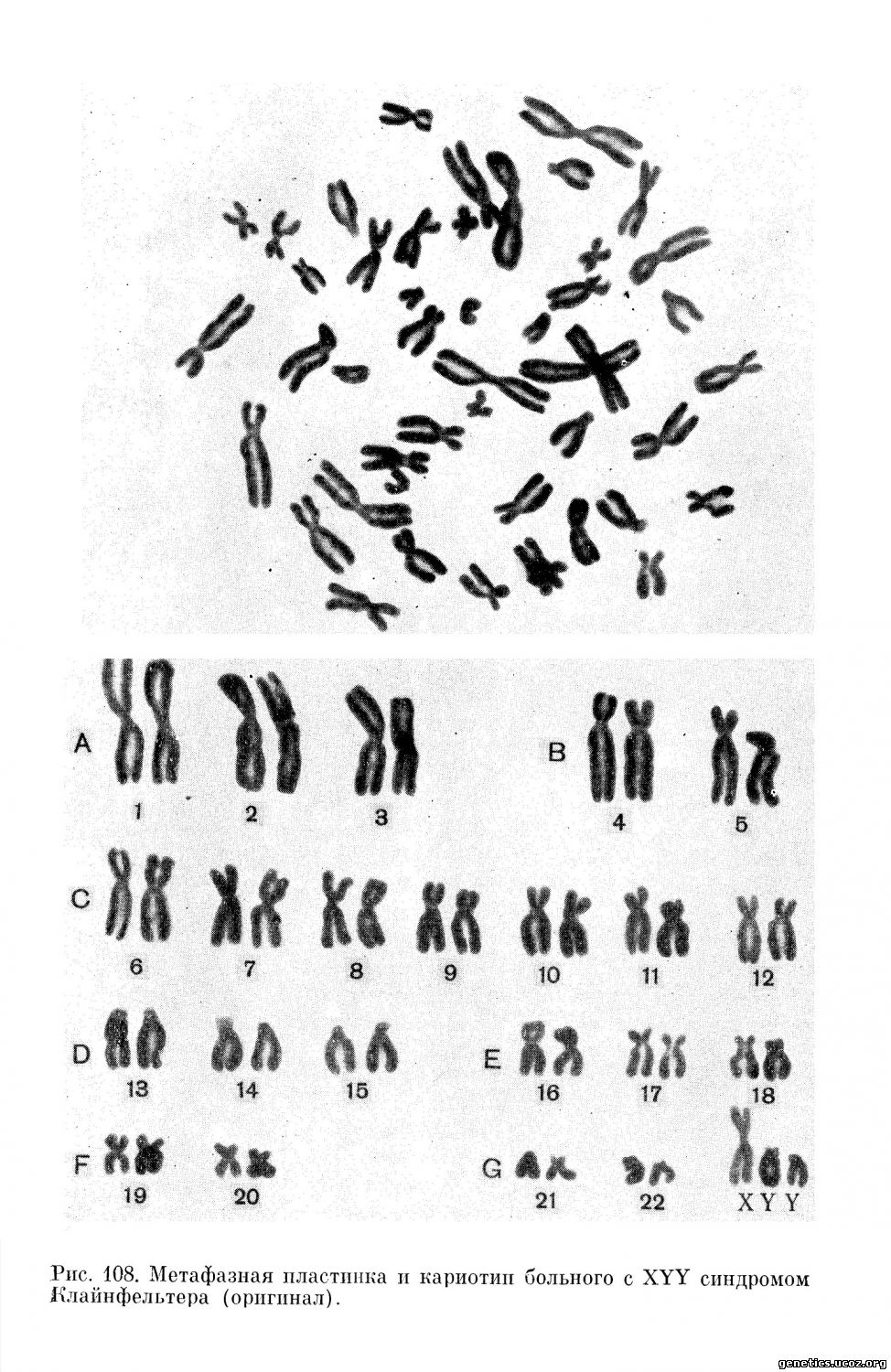
Jacobs с сотрудниками (1965), исследуя хромосомы у 197 психически больных пациентов с агрессивным поведением, в 7 случаях обнаружили XYY половой хромосомный комплекс. Интересно отметить, что средний рост у выявленных больных составлял 182,5 см (что значительно превышает норму для лиц этой возрастной группы). Высокий рост, евнухоидные пропорции тела, увеличение нижней челюсти часто отмечаются у индивидуумов с 47, XYY вариантом синдрома Клайнфельтера.
Таким образом, имея некоторые общие черты с полисомным по X-хромосоме вариантом, 47, XYY вариант синдрома более благоприятен и мягок в своих клинических проявлениях. Значительная часть таких индивидуумов выпадает из поля зрения врачей ввиду своей полноценности.
Описано несколько случаев этого синдрома с гипогонадизмом (Fraccaro и др., 1962; Milcu и др., 1964) и задержкой умственного развития (Fraccaro и др., 1962).
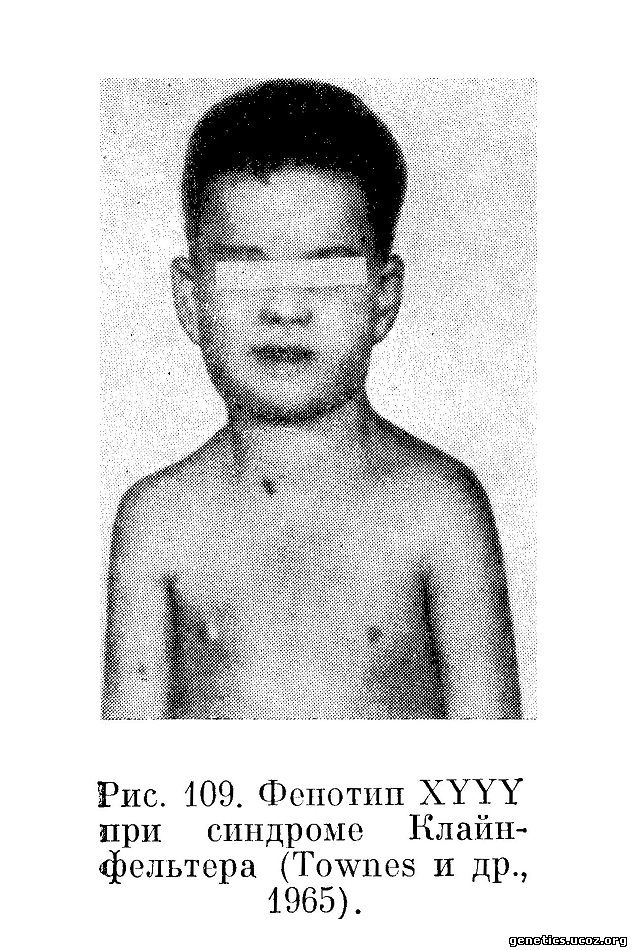
Единственного 48, XYYY индивидуума описал Townes с соавторами (1965). Это был 5-летний мальчик с некоторой психомоторной задержкой. Справа в мошонке у него было нормальное яичко, в то время как слева яичко отсутствовало. Отмечалась гиперэкстензия коленных и локтевых суставов, стеноз легочного клапана. Его двоюродная сестра со стороны матери страдала болезнью Дауна — мозаик трипло-21 (рис. 109 и 110).
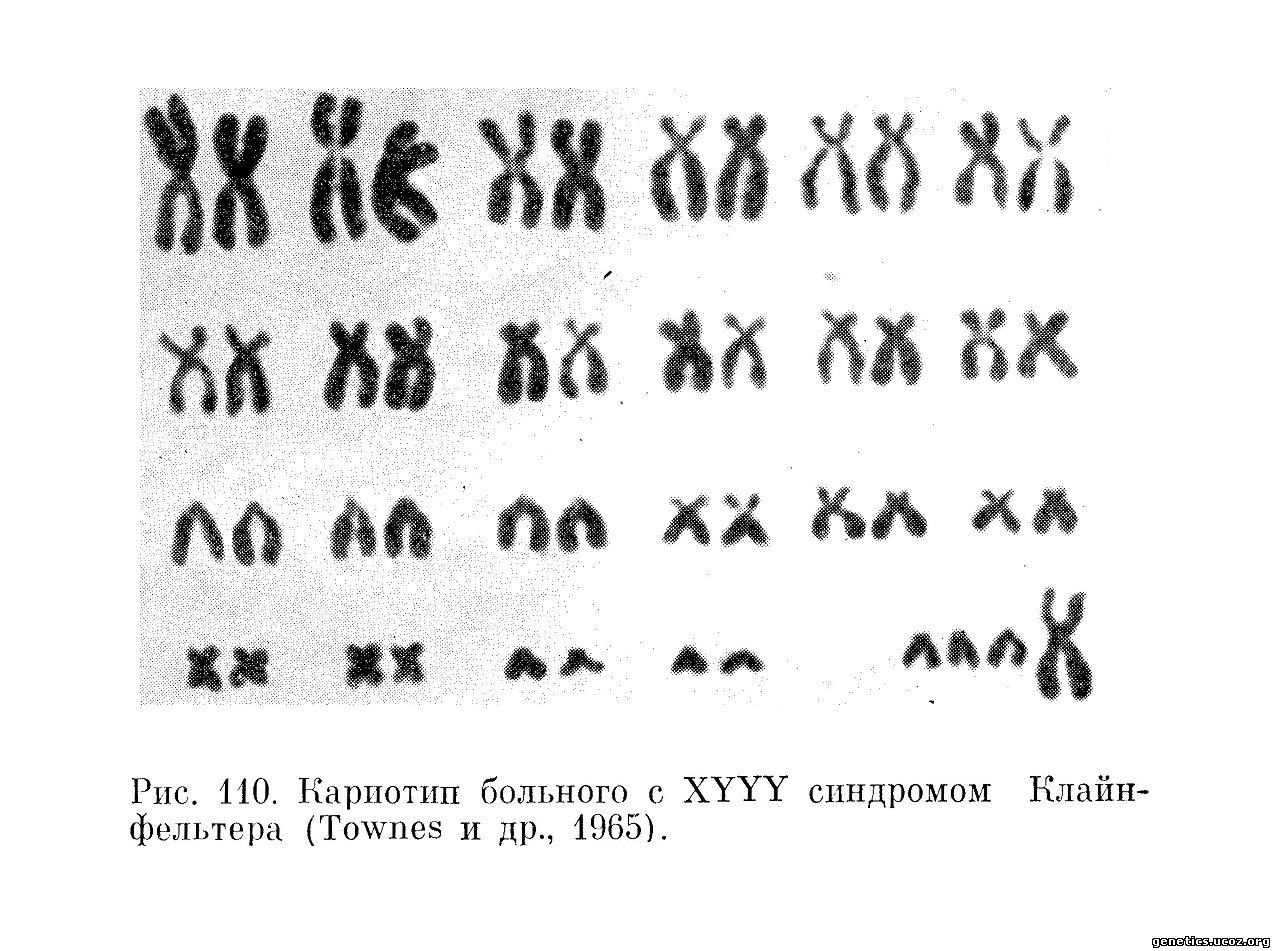
Вариант синдрома Клайнфелътера, полисомный как по Х-, так и по Y-хромосомам
Впервые 48, XXYY кариотип описал Muldal с соавторами в 1962 г. у 15-летнего хроматинположительного мальчика с гинекомастией и значительным умственным недоразвитием. Рост его был 168 см, пропорции тела евнухоидные, мышечный тип телосложения; оволосение в подмышечных впадинах отсутствовало, на лобке отмечалось умеренное оволосение по женскому типу; двусторонний equinovarus. Рентгенологически обнаруживались двусторонние цервикальные ребра. Половой член нормальный, яички уменьшены в размерах. Гистологическая картина напоминала изменения, обнаруживаемые в постпубертатных яичках больных с 47, XXY синдромом Клайнфелътера.
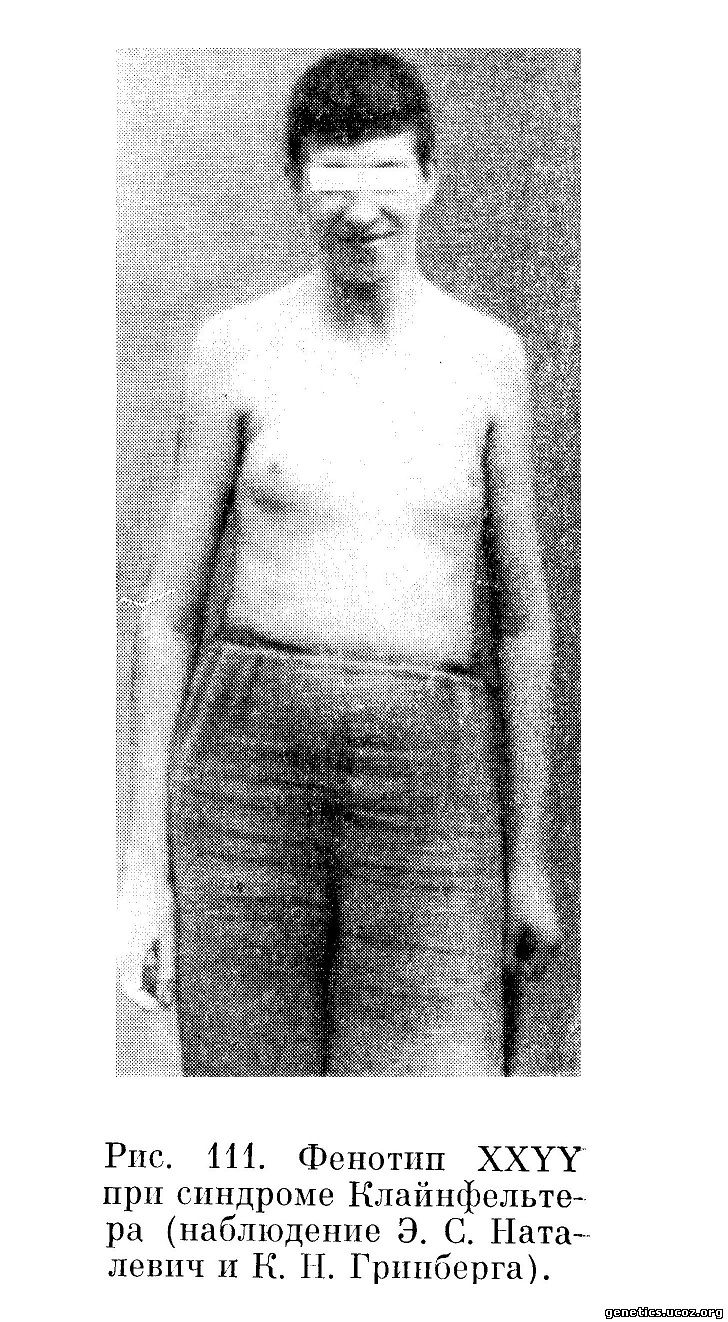
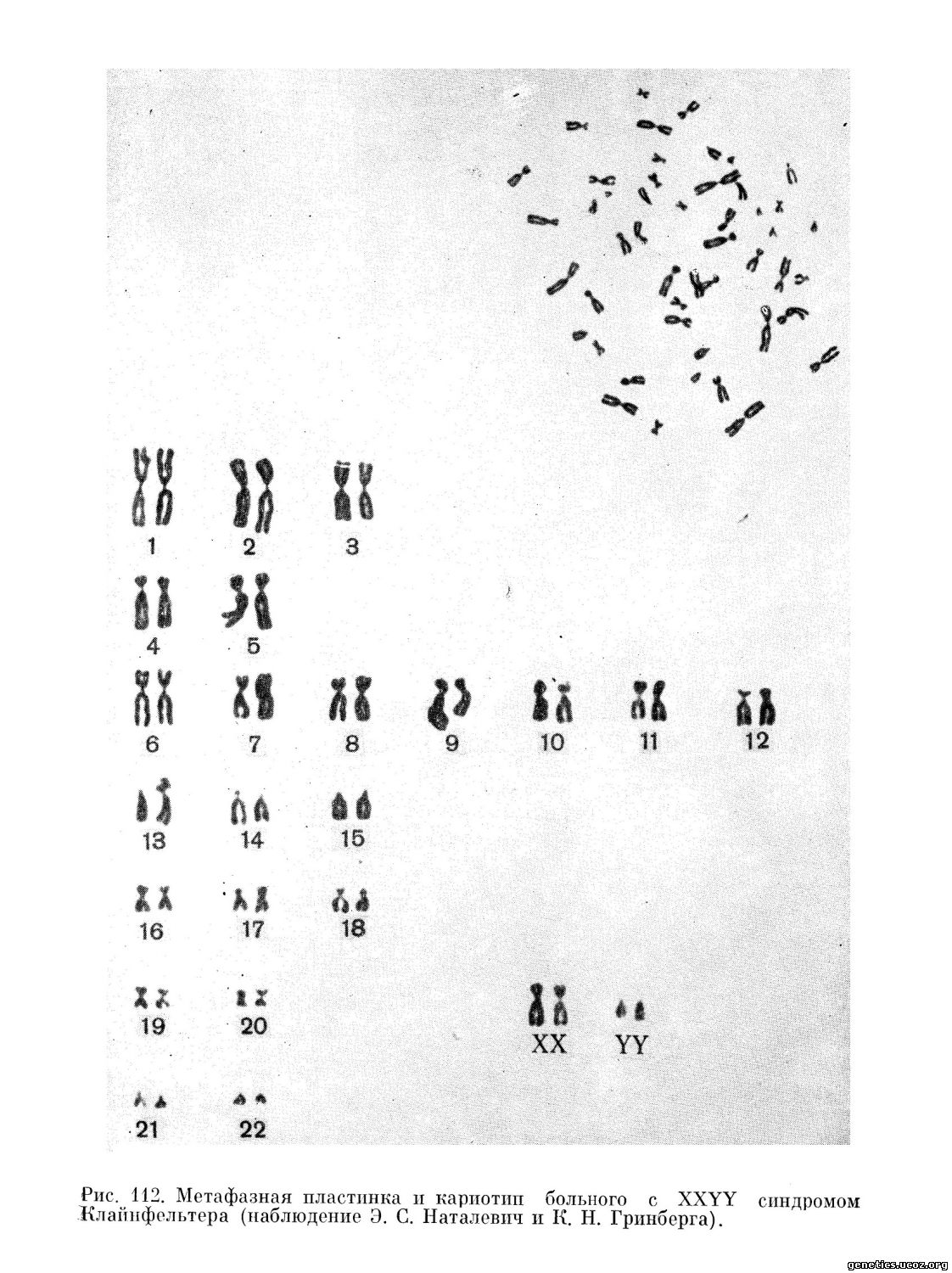
В ряде случаев, описанных в дальнейшем (Leisti и др., 1964; Schlegel и др., 1965; Waterman и др., 1966), наряду с чертами, общими для 47, XXY варианта, отмечалась тенденция к гигантизму и проявлению акромегалоидных черт: увеличение фронтальных пазух, выдающийся подбородок. Иногда встречаются аномалии в развитии скелета. У взрослых индивидуумов часто наблюдаются варикозное расширение вен и трофические язвы нижних конечностей (рис. 111 и 112).
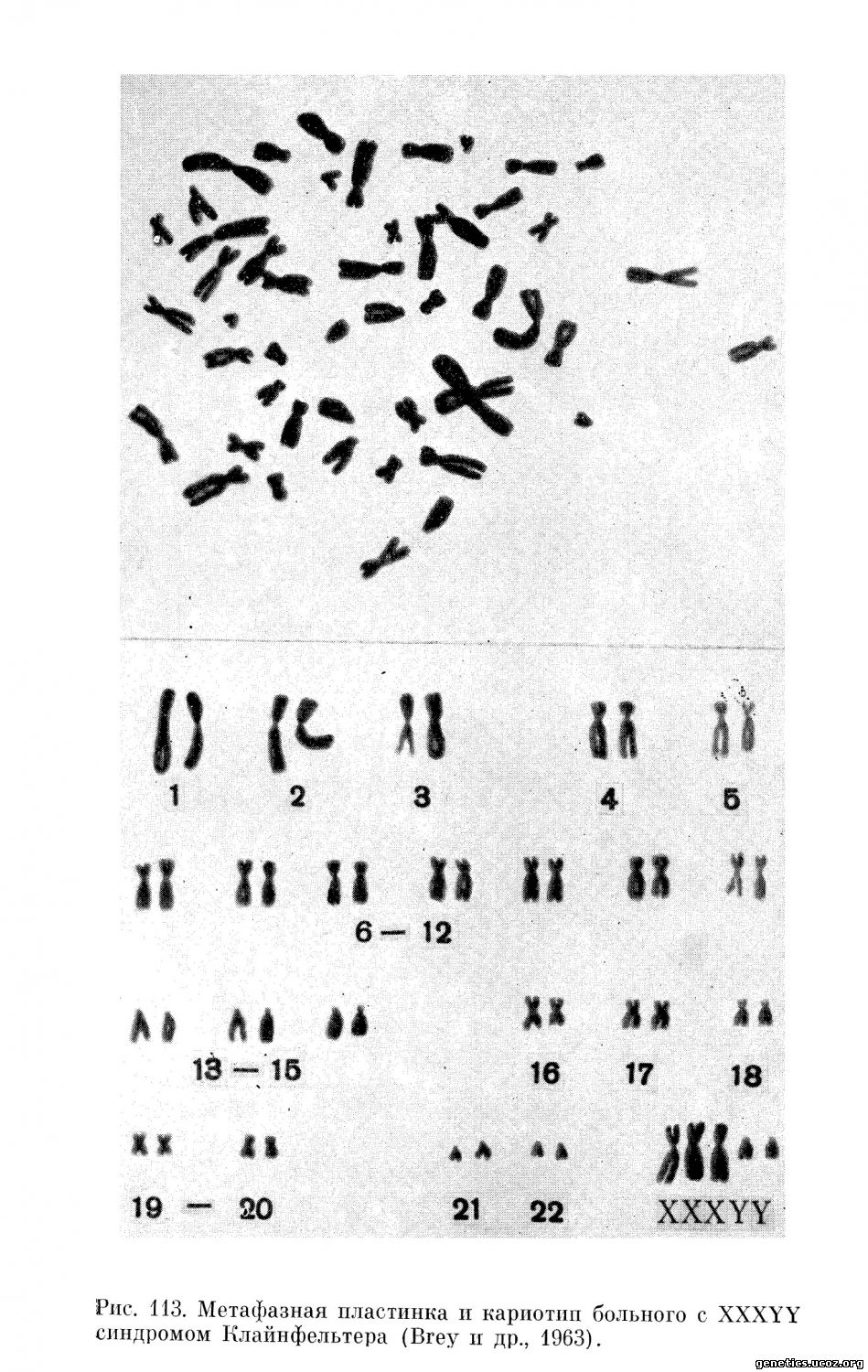
Единственный 49, XXXYY кариотип (рис. 11З) был описан Bray и Josephine в 1963 г. у 25-летнего мужчины с умственной отсталостью. Пациент был высокого роста с евнухоидными пропорциями тела. Оволосение на лобке, в подмышечных впадинах и на лице отсутствовало, имелась гинекомастия и увеличение нижней челюсти. В маленьких атрофичных яичках при гистологическом исследовании были обнаружены только элементы стромы. Семенные трубочки и клетки Лейдига отсутствовали, отмечались «тени канальцев», представляющие собой остатки семенных трубочек.
Мозаицизм при синдроме Клайнфелътера
Наряду с описанными выше имеется еще множество вариантов синдрома Клайнфельтера, при которых кариологический анализ обнаруживает два и более клеточных клонов — «мозаичные варианты синдрома».
Вскоре после выявления XXY конституции по половым хромосомам при синдроме Клайнфельтера Ford с сотрудниками (1959), описали 46, ХХ/47, XXY вариант этого синдрома, а в дальнейшем подобные индивидуумы были обнаружены многими исследователями (Hayward, 1960; Nowakowski и др., 1960). Являясь хроматинположительными, клинически эти пациенты почти не отличаются от «чистых», немозаичных 47, XXY индивидуумов.
XY/XXY — наиболее частый тип мозаичности по половым хромосомам у мужчин (Maclean и др., 1964). Такие «мозаики» чаще всего хроматинположительные (Maclean и др., 1961; Wright и др., 1963; Warburg, 1963). У большинства хроматинотрицательных больных с синдромом Клайнфельтера обнаруживается 46, XY кариотип (Court Brown и др., 1960). Однако у некоторых больных с отсутствием телец Барра в соскобе со слизистой оболочки полости рта были найдены как 46, XY, так и 47, XXY клеточные линии (Sandberg и др., 1960), что предполагает хромосомную этиологию у большинства индивидуумов с этим синдромом.
Присутствие нормального мужского (46, XY) клеточного клона смягчает тяжесть проявления этого синдрома. У пациентов с такой мозаикой часто имеется меньшая степень атрофии тестикул, у некоторых из них сохраняется сперматогенез (Ferguson-Smith и др., 1957; Raboch, Bleha, 1960). Описаны отдельные индивидуумы с синдромом Клайнфельтера, имеющие детей (Warburg, 1963).
Следует ожидать, что пациенты с 46, XY/47, XXY мозаичностью должны встречаться значительно чаще, чем «чистые» формы этой аномалии полового развития. Многие из них, вероятно, будучи нормальными в половом отношении, не обращаются за медицинской помощью и ускользают из поля зрения исследователей.
Описаны 45, Х/46, XY/47, XXY (Miller, 1962), 48, XXXY/49, XXXXY (Jacobs и др., 1961), 47, ХХХ/49, XXXXY (Stimson и др., 1963), 48, XXXY/49, XXXXY/50, XXXXXY (Anders и др., 1960) «мозаики», мало отличающиеся по клинической картине от соответствующих «чистых» форм этого синдрома.
Изменение структуры Х-хромосомы
Структурные изменения Х-хромосомы не вносят каких-либо заметных отклонений в клиническую картину синдрома Клайнфельтера.
Делеция значительной части длинного плеча одной из Х-хромосом обнаружена Crawfurd (1961) у мозаичного индивидуума с хроматинположительным синдромом Клайнфельтера и наличием врожденных аномалий костной системы. Делетированная хромосома напоминала хромосому 18.
47, XXq — Y/46, XY мозаицизм у хроматинположительного умственно отсталого больного с признаками синдрома Клайнфельтера описал Neilsen (1966). Делетированная Х-хромосома в этом случае напоминала по форме хромосому 16.
Делеция короткого плеча Х-хромосомы с признаками синдрома Клайнфельтера обнаружена у хроматинотрицательного 46, XY/47, ХХр — Y/46, ХХр «мозаика». Делетированная хромосома не отличалась от больших акроцентрических хромосом (Valencia и др., 1964).