В 1960 г. Edwards с сотрудниками сообщил о новом синдроме, связанном с трисомией по группе Е. Они исследовали девочку, извлеченную путем кесарева сечения. Ее родители (мать 31 года и отец 32 лет) были здоровы, так же как и ее 6-летний брат.
До рождения этой девочки у матери было два выкидыша. Беременность протекала с токсикозом и отмечалось многоводие.
У ребенка наблюдались следующие особенности: голова необычной формы с узким фронтальным и широким затылочно-теменным диаметром, широкая спинка носа, низко расположенные уши, маленький треугольный рот, затруднявший кормление грудью, перепонки между пальцами. Отмечалась чрезмерная подвижность плечевых суставов и короткопалость. У девочки прослушивался выраженный систолический шум в четвертом межреберье. В первые недели жизни отмечались преходящая желтуха, которая была оценена как обтурационная, и приступы цианоза. Ребенок был помещен в детскую клинику, где подвергся тщательному исследованию. Девочка явно отставала в развитии: к 2 месяцам она не улыбалась, не могла фиксировать взгляд, крик был очень слабый. Отмечалось увеличение печени. Наружные гениталии были нормальные. Моча и кровь без изменений. Рентгенологически установлено увеличение сердца. Биопсия печени и диагностическая лапаротомия показали наличие гигантоклеточного гепатита, интерстициального фиброза и желчных тромбов в центральных долях печени. Произведена операция холецистоеюностомии. После операции возникло кровотечение, от которого ребенок умер, несмотря на переливание крови. Для цитогенетического исследования были взяты кусочки кожи и мышц спустя 3 часа после смерти. Получена культура, и в клетках найдено 47 хромосом. Лишняя хромосома относилась к группе Е, и авторы полагали, что это хромосома из пары 17. Таким образом, был обнаружен новый синдром, связанный с трисомией по группе Е (47, XX, Е + ).
Вскоре были опубликованы сообщения с описанием таких же случаев (Patau и др., 1960, 1961). К настоящему времени описано уже около 100 случаев этого заболевания.
а) Цитогенетика
Хромосомы группы Е представляют собой субметацентрические хромосомы средних размеров. Patau и Yunis (1965) и В. М. Гиндилис (1967) считают, что хромосомы этой группы достаточно морфологически отличаются друг от друга, чтобы точно их идентифицировать. Хромосома 18 более субметацентрична, чем 17 и особенно 16. В отличие от хромосом 16 и 17 на хромосоме 18 не отмечаются или отмечаются очень редко вторичные перетяжки (рис. 136). Авторадиографические исследования (Yunis, 1965) показали, что в группе Е наиболее поздно метящимися являются хромосомы 18, причем в случаях синдрома, о котором идет речь, поздно метящейся является именно хромосома, вовлеченная в трисомию.
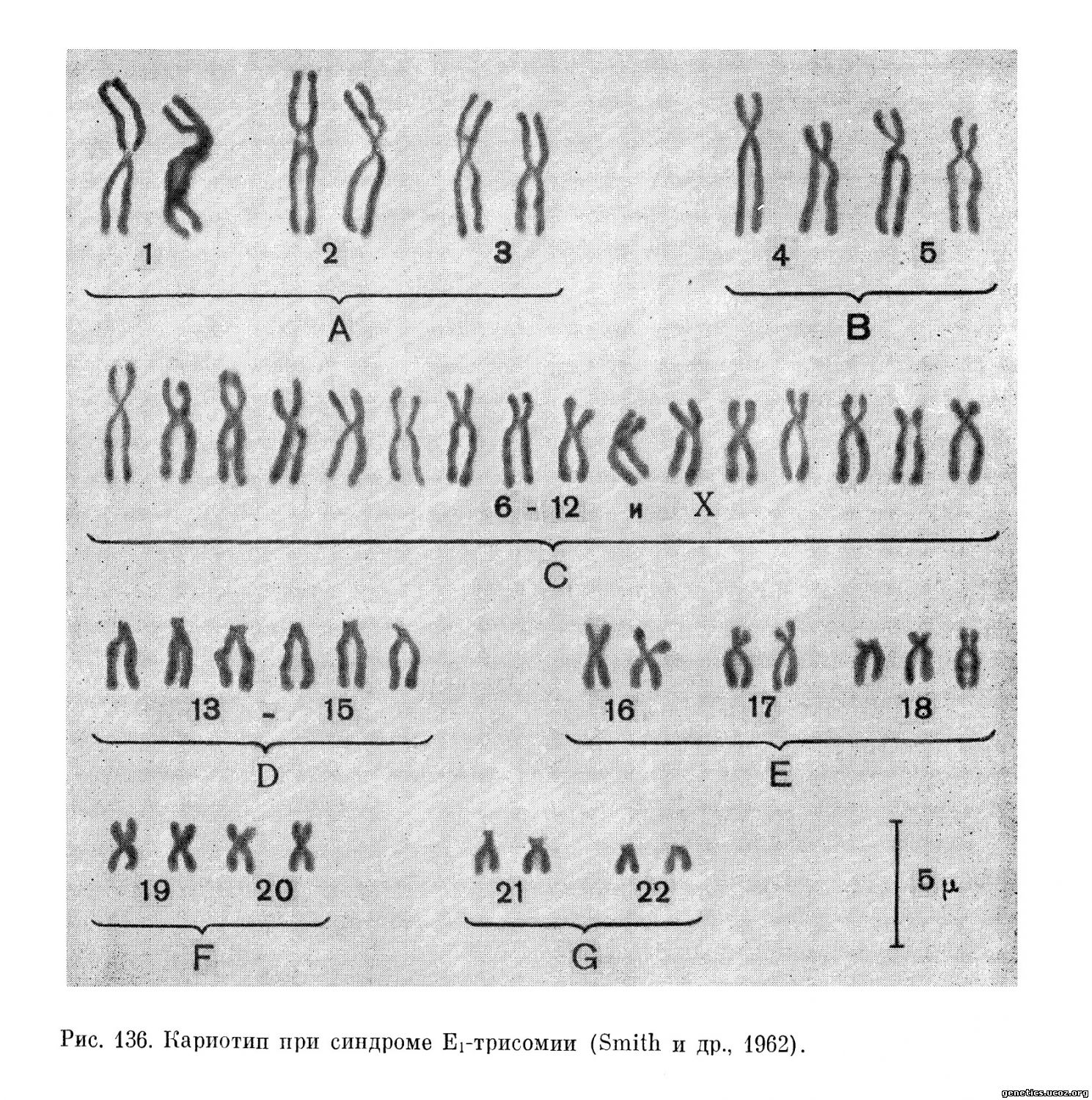
Несмотря на возможность точной идентификации, некоторые авторы, в частности Edwards (1963), настаивают на необходимости говорить не о синдроме трисомии по хромосоме 18, а о синдроме Еі-трисомии.
б) Частота заболевания
Так же как и в случае синдрома Di-трисомии, разные авторы приводят различные цифры частоты этой аномалии. Это связано либо с малым объемом выборки, либо с неслучайным распределением частоты аномалии во времени (явление «Clustering»). Так, Marden с соавторами (1964) дают цифру 1:4348 живорождений, a Hecht с сотрудниками (1963) сообщают о другой частоте—1 случай на 500 живорождений. Heinrics и др. (1963) показали реальное существование явления «Clustering» для Еі-трисомии: за 14 месяцев они обнаружили в обследуемой популяции 2 случая (1 : 118,5 живорождений), в то время как в предыдущие годы не было ни одного. Сопеп и Erkman (1966), критически рассмотрев данные литературы и присоединив свои 17 случаев, приводят среднюю частоту синдрома E-трисомии, равную 2,2*10-4 (1 случай на 4500 живорождений), причем эти авторы допускают, что 25—50% случаев в их исследовании и других наблюдениях, терялось из-за диагностических ошибок или не доходило до последовательного центра. Впервые Ferguson-Smith (1962) обратил внимание на ненормальное соотношение полов при синдромах, связанных с трисомией аутосом. Сопеп и Erkman на большом материале подтвердили это, показав, что среди больных с синдромом Еі-трисомии явно преобладает женский пол. В том же исследовании эти авторы отмечают, что в большинстве случаев больные с этим синдромом рождаются в первой половине года.
в) Возраст родителей
В большинстве работ, содержащих описание синдрома Еі-трисомии, отмечается пожилой, с акушерской точки зрения, возраст родителей. Это не касается случаев, связанных с частичной трисомией в результате транслокации. В уже приводившемся обширном исследовании Сопеп и Erkman средний возраст матерей был 31,8, отцов — 36,4 года. По-видимому, в отношении нерасхождения хромосом группы D и Е существует такая же связь между возрастом родителей и частотой нерасхождения, как и при синдроме Дауна, однако в силу большей редкости этих заболеваний трудно обнаружить столь же четкую корреляцию, как при нерасхождении хромосом группы G.
г) Наследственность
Holman с сотрудниками (1963) сообщил о семье, где 2 сибса были поражены этим заболеванием. Huggert (1966) приводит интересную родословную, где у 2 сибсов (брат и сестра) был синдром Еі-трисомии и еще у одного ребенка в предыдущем поколении также имелись клинические признаки этого заболевания. Однако этот ребенок умер еще в 1946 г. и диагноз кариологически не верифицирован. В случае, описанном Huggert, обращает на себя внимание большая частота родственных браков в роду.
В специальной работе Hecht с соавторами (1964) проанализировал данные относительно 60 семей, из которых в 44 были случаи Е-трисомии. Показано, что в семьях, где имеется синдром Дауна, частота сибсов с трисомией Еі значительно выше, чем в обычной популяции. На основании этого авторы делают вывод о неслучайном распределении хромосомных аномалий в популяции. Эта мысль подтверждается также в ряде сообщений о сочетании Еі-трисомии с аномалиями половых хромосом у одного индивидуума. Hecłit и др. полагают, что эта неслучайность может быть связана с аутоиммунными заболеваниями, мелкими структурными перестройками, ведущими к нерасхождению, с наличием специальных генов (наподобие гена claret-non-disjunction у дрозофилы) и действием вирусов (см. соответствующие разделы). Однако большинство случаев, описанных в литературе, относится к спорадическим.
Особая ситуация в отношении наследственной передачи частичной трисомии имеется в случаях транслокации.
д) Связь с заболеваниями матери и действием внешних факторов
В случае, сообщенном Uchida и др. (1962), мать пробанда долгое время работала рентгенотехником, вплоть до IV месяца беременности. Авторы полагают, что в данном случае причиной нераехождения хромосом является действие радиации.
Townes и др. (1964), проанализировав собственных 3 случая и опубликованные наблюдения, пришли к выводу, что в анамнезе родителей детей с Еі-трисомией часто можно отметить наличие диагностического рентгеновского облучения незадолго до зачатия. По мнению этих авторов, во многих случаях диагностическое облучение перед зачатием может быть причиной нерасхождения хромосом. Однако в большинстве наблюдений, описанных в литературе, не удалось найти каких-либо указаний на заболевания матери или облучение.
е) Клинические особенности синдрома
Беременность у матерей, родивших детей с синдромом Еі-трисомии, как правило, протекает нормально, однако в ряде случаев (Patau и др., 1960; Patau и др., 1961) отмечается многоводие. Продолжительность беременности обычная, но часто ей не соответствует вес больных. Более того, Townes и др. специально отметили, что чем дольше длится беременность в случаях трисомии Еі, тем меньше вес ребенка при рождении. Hecht (1963) обратил внимание, что при этом заболевании иногда бывает слишком маленькая плацента. Lewis (1964) подчеркивает, что при синдроме Еі-трисомии часто находят единственную пупочную артерию. Lenosky, Medovy (1962), собрав большой материал, касающийся случаев с единственной пупочной артерией, пришли к выводу, что эта аномалия часто сочетается с врожденными уродствами, в частности с теми, которые вызваны хромосомными аберрациями. Lewis считает этот признак важным для того, чтобы заподозрить трисомию. Как правило, больные дети рождаются в состоянии асфиксии, хотя роды проходят благополучно. Во всех случаях отмечается цианоз. Внешний вид больных настолько характерен, что позволяет диагностировать Еі-трисомию до цитологического исследования, которое подтверждает диагноз в большинстве случаев (Соnеn, Erkman, 1966) (рис. 137).

Череп необычной формы — узкий лоб и широкий выступающий затылок. В ряде случаев отмечается гидроцефалия (El-Alfi и др., 1964). Весьма характерно расположение ушных раковин: очень низко, на уровне углов рта. Наружное ухо сформировано неправильно: отсутствует мочка и козелок, недоразвит завиток и противозавиток (рис. 138).
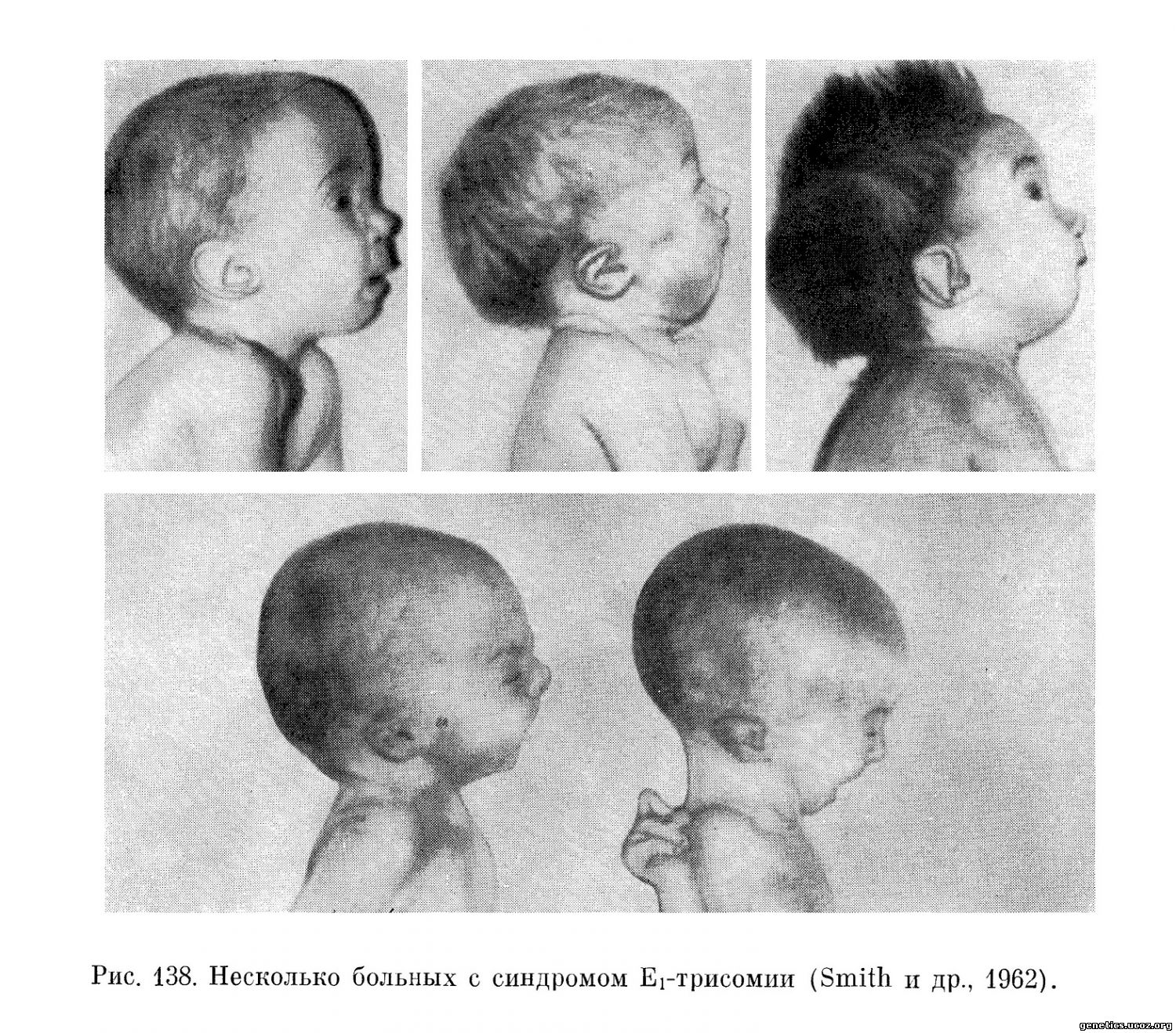
Постоянным признаком является микрогнатия, особенно гипоплазия нижней челюсти. В результате этого рот очень маленький, треугольной формы. Это обстоятельство часто препятствует кормлению грудью. Весьма характерный вид имеют кисти (рис. 139, 140), пальцы широкие и короткие. Наблюдается гипоплазия мышц возвышения большого пальца. Пальцы кисти сложены особым образом, что обозначается термином «флексорная аномалия пальцев». Средний и безымянный пальцы согнуты и приведены к ладони, мизинец и указательный накладываются на них сверху. На ладонной поверхности кисти постоянно находят поперечную складку («обезьяньяскладка»).
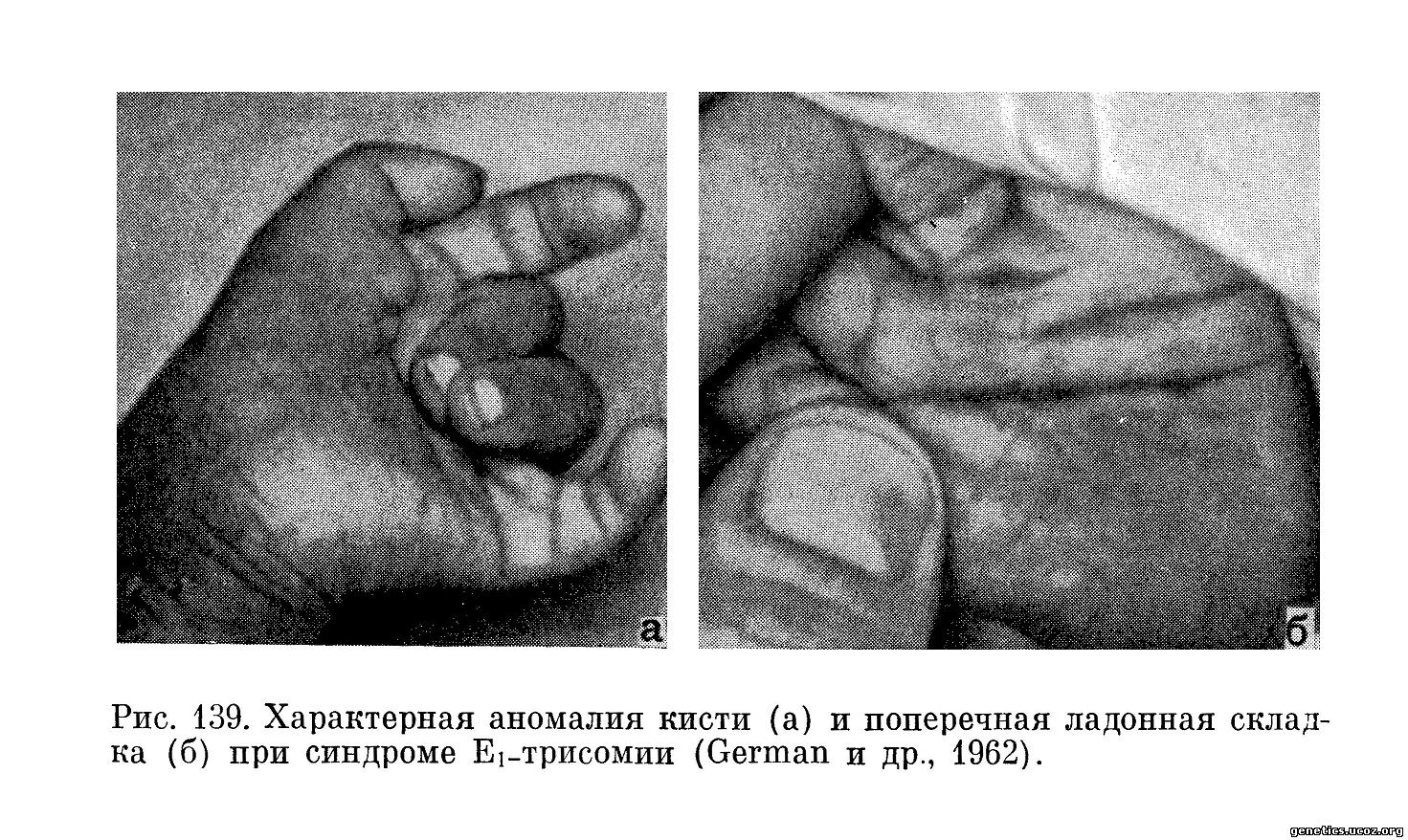

Подвижность в тазобедренных суставах ограничена, бедра в состоянии аддукции. Большой палец стопы находится в дорсальной флексии (рис. 141).
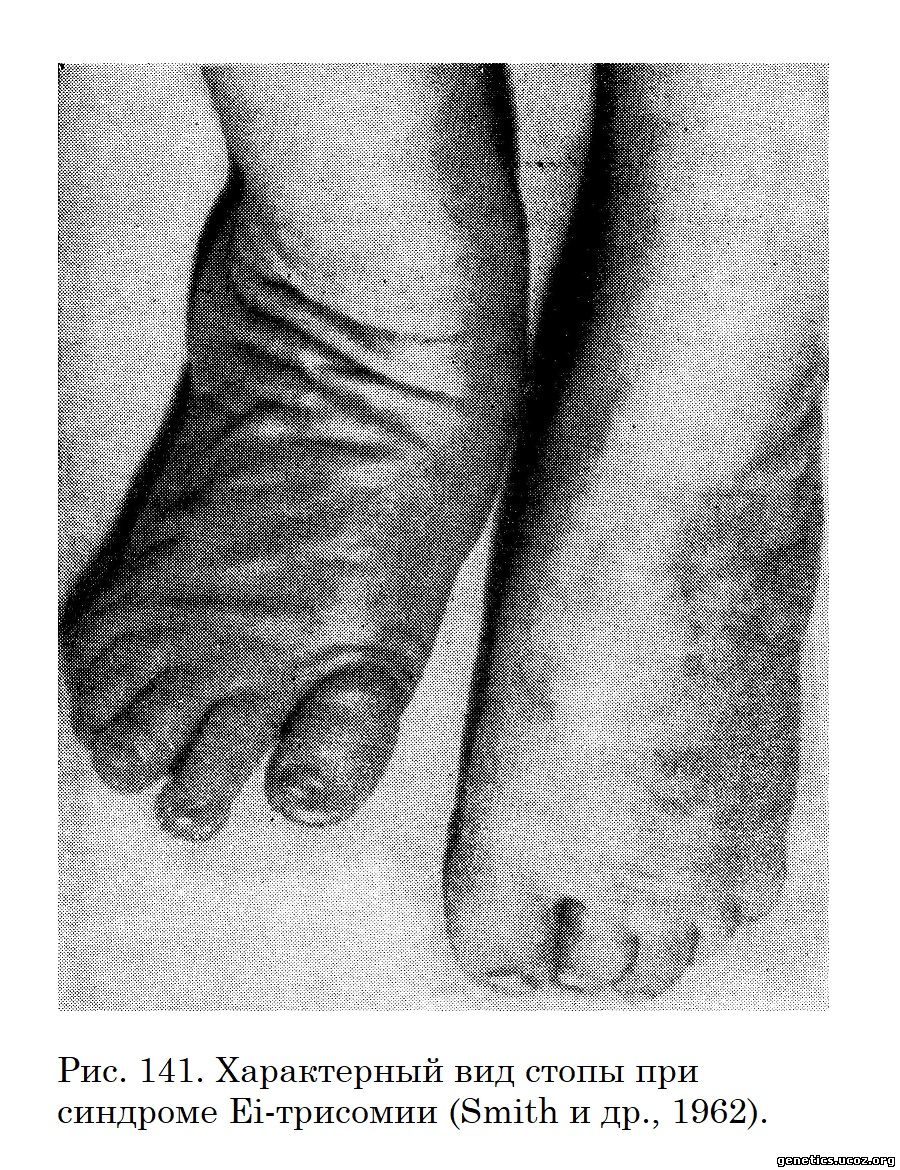
Постоянно обнаруживаются аномалии формирования стопы, что выражается в talipes equinovarus («rocker-bottom feet»). Грудная клетка широкая, расстояние между сосками большое. Характерно укорочение грудины. Таз узкий. Часто отмечается синдактилия. Кожа чрезмерно подвижна и часто образует характерные складки на шее. Подкожножировая клетчатка развита слабо, иногда почти отсутствует. Довольно часто бывает односторонний или двусторонний птоз и эпикант. Отмечается общая гипоплазия мышц или отдельных групп и мышечная гипотония. Не постоянно, но достаточно часто находят помутнение роговицы (Huggert, 1966; Townes и др., 1962). Во всех случаях имеются клинические признаки врожденного порока сердца.
Дети с такими аномалиями рождаются весьма слабыми, малоактивными. Чрезвычайно характерно отсутствие прогредиентности в их развитии, они не прибавляют в весе, психомоторное развитие тормозится. В литературе это обозначается термином «failure to thrive» и является патогномоничным симптомом (рис. 142).
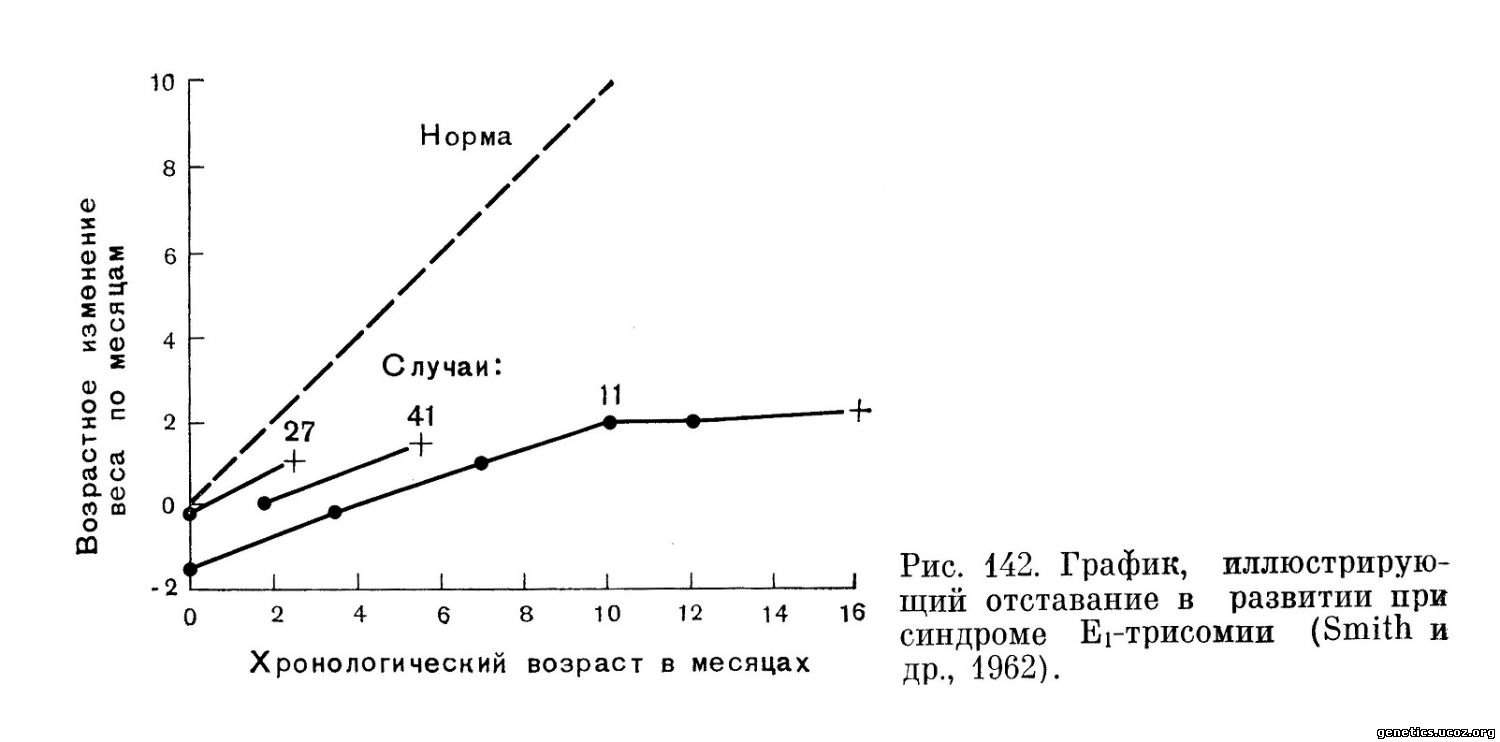
Крик у детей очень слабый, сосательный рефлекс слабо развит. Часто отмечаются приступы цианоза и желтухи. Нередко судорожные припадки. Дети умирают, как правило, от сердечной недостаточности или от присоединившейся инфекции, чаще всего поражающей легкие (бронхопневмония) или почки (пиелонефрит). Жизнеспособность больных резко снижена. Среднее время выживания для мальчиков — 58 1/2 дней, для девочек — 282 дня (Сопеп, Erkman, 1966). С лучшей выживаемостью девочек связано и преобладание женского пола в случаях Еі-трисомии. Следует отметить, что не все перечисленные признаки встречаются достаточно часто сразу у одного индивидуума. При синдроме Еі-трисомии, так же как и при синдромах, связанных с аномалиями других хромосом, нет специфических симптомов (рис. 143).

Все указанные аномалии, изолированные, либо в том или ином сочетании, встречаются при разных заболеваниях. Синдром Еі-трисомии, как и синдром Di-трисомии, синдром Дауна и синдром делеции короткого плеча одной из хромосом группы В, характеризуется специфическим сочетанием отдельных аномалий. Одни из этих аномалий являются обязательными, наблюдаются постоянно, другие — встречаются от случая к случаю.
Наиболее подробно проанализировал патологию Е-трисомии Lewis (1964). На основании изучения собственных 7 случаев и данных литературы он выделяет постоянные симптомы, встречающиеся более чем в 50% случаев. Это следующие признаки (в порядке убывания частоты): низкое расположение ушей, маленькая челюсть, клинические симптомы порока сердца, неправильно сформированные ушные раковины, флексорная аномалия пальцев, короткая грудина, характерное наложение пальцев друг на друга, выступающий затылок, высокое небо. К обычным, но непостоянным аномалиям, наблюдающимся в 10—50% случаев, относятся (в порядке убывания частоты): ограничение подвижности в тазобедренном суставе, дорсальная флексия большого пальца стопы, широкая грудная клетка, обезьянья складка, кожные складки на шее, гипоплазия мышц, короткий большой палец, синдактилия, стопа в виде «качалки», маленький рот, маленький таз, необычная форма черепа, птоз, крипторхизм, эпикант, рудиментарные ногти, маленькие губы, большой клитор, помутнение роговицы.
ж) Патологическая анатомия
На аутопсии находят множество пороков развития, затрагивающих различные органы и системы. Нередки находки, связанные со вторичными патологическими процессами, развившимися на фоне врожденных уродств.
Сердце. Как уже указывалось, постоянным признаком Е-трисомии являются клинические симптомы врожденного порока сердца. Этому соответствуют и патологоанатомические находки. Размеры и вес сердца увеличены, особенно правого желудочка.
Всегда отмечается дефект межжелудочковой перегородки, незаращение овального отверстия и открытый артериальный проток. В ряде случаев обнаружены аномалии строения клапанов. Scarpa, Borgaonkar (1966) описали дефект нижней части межпредсердной перегородки и верхней части межжелудочковой перегородки, открытый артериальный проток и общий атриовентрикулярный клапан (А.—V. communis defect). Habedank (1964), Uchida и др. (1962), а также другие авторы находили коарктацию аорты. Довольно часто обнаруживается двустворчатый аортальный клапан (Zellweger и др., 1964; Gottlieb и др., 1962). Ѵоогhes и др., (1962) установили у больного тетраду Фалло.
Легкие. Особых аномалий не бывает. Как правило, отмечаются явления полнокровия, нередко бронхопневмония.
Зобная железа. Довольно часто наблюдается гипоплазия, не соответствующая возрасту (Uchida, 19626; Lewis, 1964).
Пищеварительная система. Holman и др. (1963) у одного из сибсов с Еі-трисомией обнаружили трахео-эзофагальную фистулу. Более частыми находками являются меккелев дивертикул и аномалии толстого кишечника — нефиксированная ободочная кишка и неполный поворот кишечника.
Поджелудочная железа. Почти во всех случаях сообщается об аномалиях строения поджелудочной железы. Чаще всего это выражается в эктопии ее ткани, в нарушении расположения инсулярных и ацинозных образований и в фиброзе (Smith и др., 1960; Lewis, 1964).
Почки и мочеполовая система. Аномалии почек весьма характерны для синдрома Еі-трисомии. Чаще всего находят дольчатую почку, иногда агенезию почек. Почти постоянной находкой являются микроцисты (Rohde и др., 1964). В ряде работ отмечается дупликация и дилятация мочеточников.
Heinries и Allen (1963) обратили внимание на сходство синдрома Еі-трисомии с синдромом Поттер-агенезии почек. Действительно, в фундаментальном труде Potter (I960) на рисунках, иллюстрирующих синдром агенезии почек, можно видеть некоторые признаки, характерные для Еі-трисомии.
Представляет большой интерес наблюдение Hilson (1957), основанное на анализе достаточно большого материала; автор указывает, что пороки развития ушной раковины могут часто сочетаться и служить сигналом для обнаружения пороков развития мочеполовой системы.
Мышцы и соединительная ткань. Гипоплазия мышц и отдельных мышечных групп — характерный признак синдрома Еі-трисомии. Аномалии развития мышц и соединительной ткани приводят к диастазу прямых мышц живота, дефектам диафрагмы с эвентрацией органов грудной клетки, к паховым и пупочным грыжам.
Нервная система. Voorhes и др. (1962) опубликовали случай синдрома Еі-трисомии, в котором отмечалось частичное отсутствие мозолистого тела. Это довольно частая находка при указанном заболевании.
Finley и др. (1963) сообщили об атрофии извилин, Zelweger и др. (1964) —о случаях, в которых отмечено отсутствие границ между серым и белым веществом. Passarge и др. (1966) обнаружили у 2 детей с синдромом Еі-трисомии миеломенингоцеле, отсутствие мозолистого тела, недоразвитие мозжечка и отсутствие затылочных долей.
з) Лабораторные исследования
Кровь. Аномалии в клеточном и химическом составе не найдено. German и др. (1966) провели биохимическое исследование в случае синдрома Еі-трисомии.
Были обнаружены нормальные количественные соотношения А и А2- гемоглобинов, нормальный набор изозимов лактат-дегидрогеназы. Не было выявлено новых белков или аномального наследования гаптоглобинов и гамма-глобулинов.
Nadler и др. (1966), исследовав содержание щелочной и кислой фосфатаз в лейкоцитах, галактозо-1-фосфат-уридил-трансферазы в эритроцитах» нашли, что у больных увеличивается примерно вдвое только содержание щелочной фосфатазы. То же наблюдается при болезни Дауна. В связи с этим естественно думать, что указанное повышение неспецифично для Еі-трисомии.
Имеется сообщение о сочетании синдрома Еі-трисомии с тромбоцитопенией неизвестной этиологии (Christodoulou, Werner, 1967).
Моча. При исследовании мочи обнаруживаются явления, характерные для пиелонефрита.
Рентгенологическое исследование. При рентгенологическом исследовании находят разнообразные аномалии скелета. При рентгеноскопии грудной клетки обнаруживают увеличение сердца и усиление легочного рисунка. Рентгенологические находки при этом синдроме обобщены в работе Moseeley и др. (1963).
Lewis (1964) изучил частоту различных внутренних аномалий при синдроме Еі-трисомии. Подобно аномалиям, выявляемым при клиническом исследовании, одни из них более постоянны, другие — менее, третьи же встречаются как единичные находки. Наиболее характерные аномалии следующие (в порядке убывания их частоты): высоко расположенный дефект межжелудочковой перегородки, аномалии почек, незаращение артериального протока, незаращение овального отверстия, меккелев дивертикул, ненормальное окостенение грудины, дефект диафрагмы, диастаз прямых мышц живота, нефиксированная ободочная кишка, двустворчатый клапан легочной артерии, пупочная грыжа, паховая грыжа, малый тимус, эктопия поджелудочной железы, аномалия атриовентрикулярного клапана, двойной мочеточник, сколиоз, неполный поворот кишечника, единственная пупочная артерия, гидронефроз, двустворчатый аортальный клапан, аномалия аортального бульбуса, аномалии крестца.
и) Диагноз, прогноз и медико-генетическая консультация
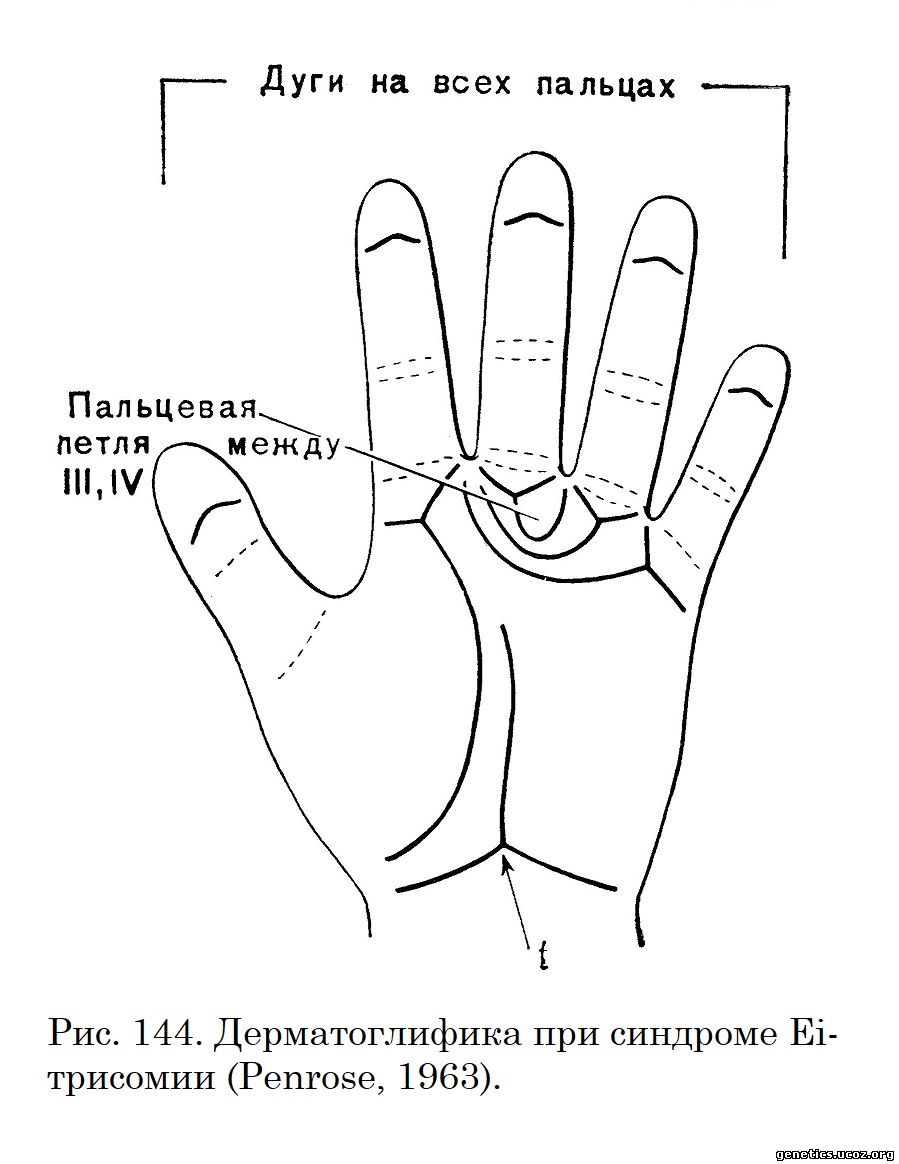
Аномалии, встречающиеся при Еі-трисомии, настолько характерны в своем своеобразном сочетании, что диагноз этой патологии возможен сразу после рождения ребенка при его осмотре. Наличие характерных дерматоглифических изменений (Uchida и др., 1962) делает диагноз почти безошибочным (рис. 144).
Важным признаком является неспособность к созреванию (failure to thrive). Цитогенетическое исследование необходимо для исключения транслокации или мозаицизма. Необходимо проводить дифференциальный диагноз с синдромом Di-трисомии. При этих заболеваниях имеются общие признаки: отставание в развитии, врожденный порок сердца, микрогнатия, низко расположенные уродливые уши, деформации конечностей. Однако как для Di-трисомии, так и для Еі-трисомии типичны определенные признаки: для Di-трисомии — микрофтальмия, анофтальмия, колобома, расщепление верхней губы и неба, полидактилия, глухота, для Еі-трисомии характерное лицо, флексорная аномалия пальцев, своеобразное строение стопы, синдактилия, короткая грудина, грыжи.
Как уже указывалось, продолжительность жизни больных невелика. Методов лечения не существует. В некоторых случаях производят операцию для ликвидации обтурационной желтухи.
Принципы медико-генетической консультации такие же, как и при синдроме Di-трисомии. Обязательным является цитогенетическое исследование для исключения транслокации и мозаицизма. Нужно учитывать несколько большую частоту синдрома Еі-трисомии, чем Di-трисомии, а также повышение частоты рождения больных детей с увеличением возраста матери.
к) Нетипичные случаи, связанные с мозаицизмом и сочетанием с другими хромосомными аномалиями
Мозаицизм
Warkany и др. (1964) сообщили о 8-летней умственно отсталой девочке с внешними аномалиями, у которой 80% клеток в культуре лейкоцитов периферической крови содержали лишнюю хромосому группы Е. Koulischer и др. (1963) описали семью, в которой у одного ребенка, умершего в возрасте 11 месяцев, имелся мозаицизм по Еі-трисомии. В отличие от типичного синдрома отмечались значительные изменения нервной системы — недостаточная миелинизация, астроцитарная пролиферация, фибриллярный глиоз. Брат и сестра этого ребенка умерли раньше и у них имелись такие же аномалии, но кариотип их и родителей не исследовался.
Weiss и др. (1962) по клиническим данным выявили 5 больных с признаками Еі-трисомии, из которых один оказался мозаиком. В случаях мозаицизма обнаруживается широкий размах вариаций в частоте отдельных симптомов. Некоторые основные симптомы могут отсутствовать. Бывают также случаи, когда все основные симптомы имеются, но цитогенетический анализ выявляет нормальный кариотип. Hook и Lewis (1965) при клиническом исследовании обнаружили у больного все характерные аномалии, однако исследование хромосом в клетках крови, кожи и костного мозга показало нормальный кариотип. Авторы полагают, что это может быть связано либо со скрытым мозаицизмом (клетки с ненормальным набором хромосом находятся в тканях, недоступных для исследования), либо с частичной трисомией в результате не обнаружимой под микроскопом инсерции. Они не исключают также возможности врожденного синдрома, обусловленного менделирующим геном, или фенокопии. Paulson, Scarborough (1966) также сообщили о больном 29 лет с некоторыми признаками трисомии и нормальным кариотипом.
Имеется ряд наблюдений над больными с сочетанием трисомии по хромосомам группы Е и с трисомией Х-хромосомы (Uchida и др., 1962; Ricci, Borgatti, 1963) (рис. 145). У таких больных сочетаются признаки Еі-трисомии и синдрома трипло-Х. У них часто находят аномалии развития половых органов. Ferguson, Pitt (1963) опубликовали случай сложного мозаицизма (Еі-трисомия + ХХХ/Еі-эусомия+ХХХ).
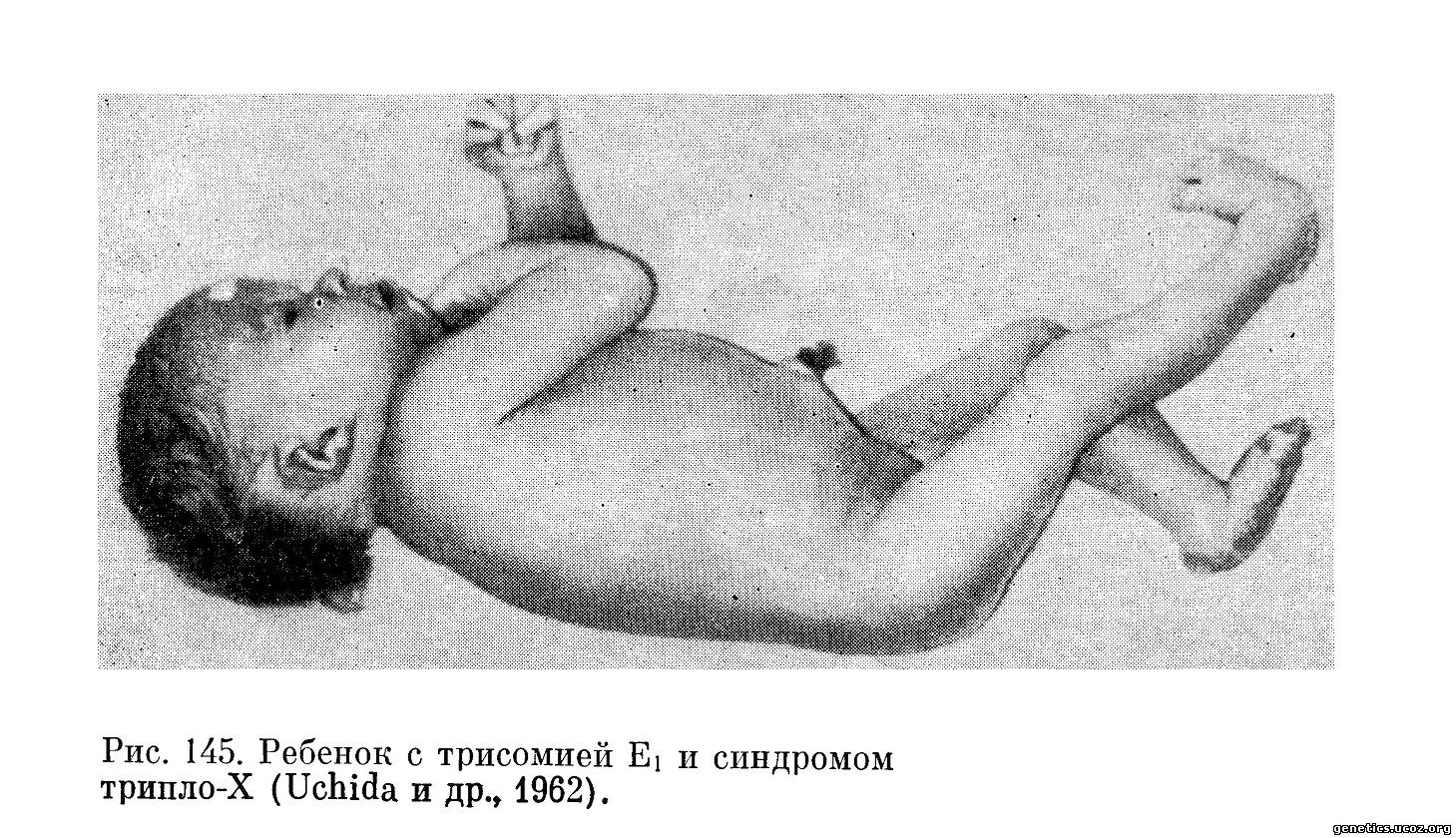
Больную с синдромом трипло-Х и Еі-трисомией описали также Haas и Lewis (1966). Во всех перечисленных случаях предварительный диагноз синдрома трипло-Х ставился на основании наличия в соскобе слизистой оболочки рта клеток, содержащих два тельца полового хроматина. Имеется также сообщение Gagnon и др. (1961) о больном, у которого одновременно имелась Gi-трисомия и Еі-трисомия. У него были признаки как синдрома Дауна, так и синдрома Еі-трисомии.
Совпадение таких достаточно редких явлений, как нерасхождение по двум хромосомам, заставляет думать о наличии общей для них причины.