Хотя первичные делеции — нередкая хромосомная аберрация у человека, в литературе встречаются главным образом описания больных, имеющих делеции по хромосомам некоторых определенных групп или даже пар, и почти совершенно не описаны делеции в хромосомах других групп или пар. Чаще других встречаются описания делеции короткого плеча хромосомы из группы В (синдром «кошачьего крика»), делеция длинного и короткого плеча хромосомы 18, делеции длинного плеча хромосом из группы D и Е. Очень часты упоминания о делеции в Х-хромосоме. Чрезвычайно редко встречаются описания делеций по другим хромосомам, а по некоторым парам нет ни одного сообщения. Это может быть связано как с истинной частотой этих событий, так и с различной выживаемостью больных с нехваткой материала из той или иной хромосомы.
а) Синдром «кошачьего крика»
В 1963 г. Lejeune и 6 его сотрудников описали 3 больных с новым хромосомным заболеванием, которое они предложили назвать синдромом «кошачьего крика» (Cri du chat). Своим необычным названием этот синдром обязан характерному крику, который издают до определенного возраста больные дети. К настоящему времени уже опубликовано значительное число наблюдений этого синдрома. Помимо своеобразного крика, напоминающего кошачье мяуканье и обусловленного аномалиями развития гортани, эта болезнь характеризуется целым рядом симптомов (рис. 150).
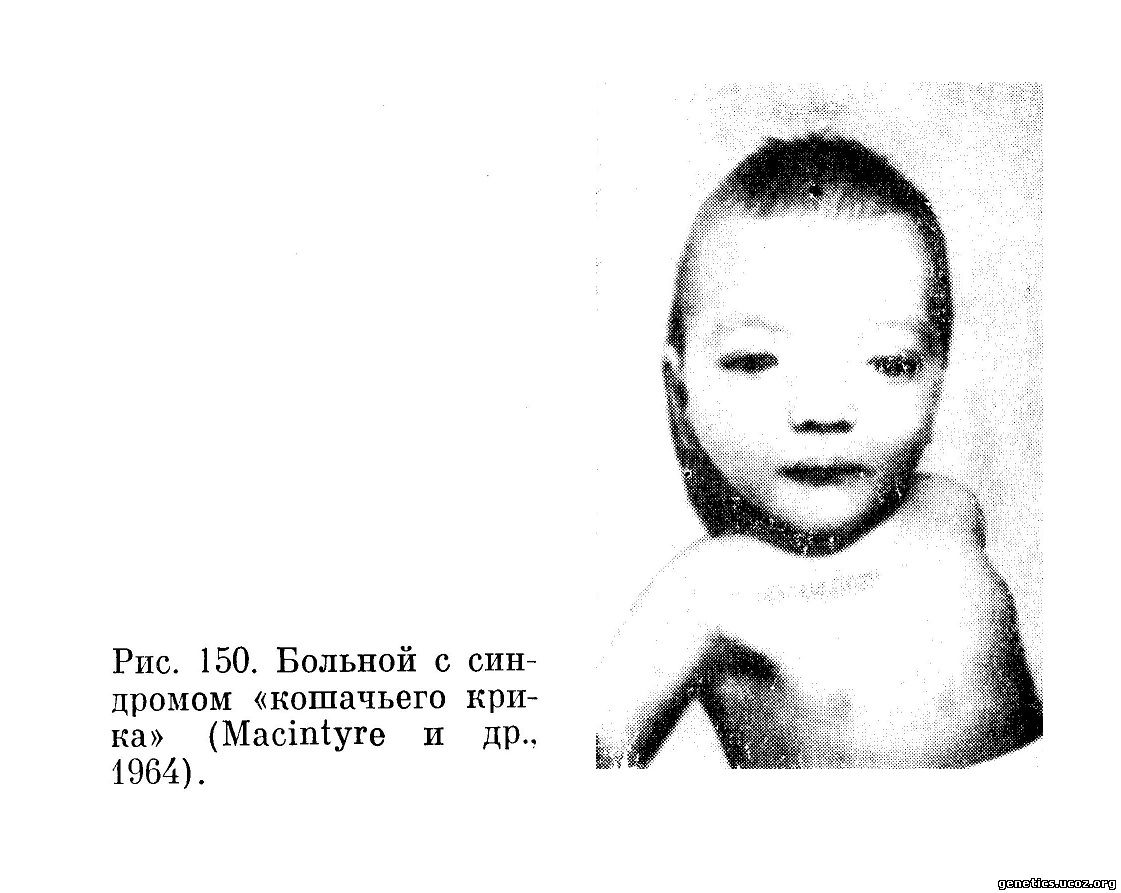
Из них наиболее характерны тяжелая умственная отсталость, задержка роста и развития, круглое, лунообразное лицо, микроцефалия, микро- и ретрогнатия, гипертелоризм, антимонголоидный разрез глаз, эпикант, низко расположенные дисплаотичные уши, мышечная гипотония, иногда грыжи, а в некоторых случаях отмечены парезы верхних и нижних конечностей, гипоплазия половых органов и слабая выраженность вторичных половых признаков; характерные для хромосомной патологии изменения рисунка кожных складок на ладонях, иногда короткая шея, но без pterigium coli. Вскоре после первого сообщения Lejeune с сотрудниками появилось большое число работ, посвященных этому заболеванию (Lejeune и др., 1964; Macintype и др., 1964; Dumars и др., 1964).
Во всех случаях у больных находили нехватку значительной части короткого плеча одной из хромосом группы В (46, XX, Вр—) (рис. 151).
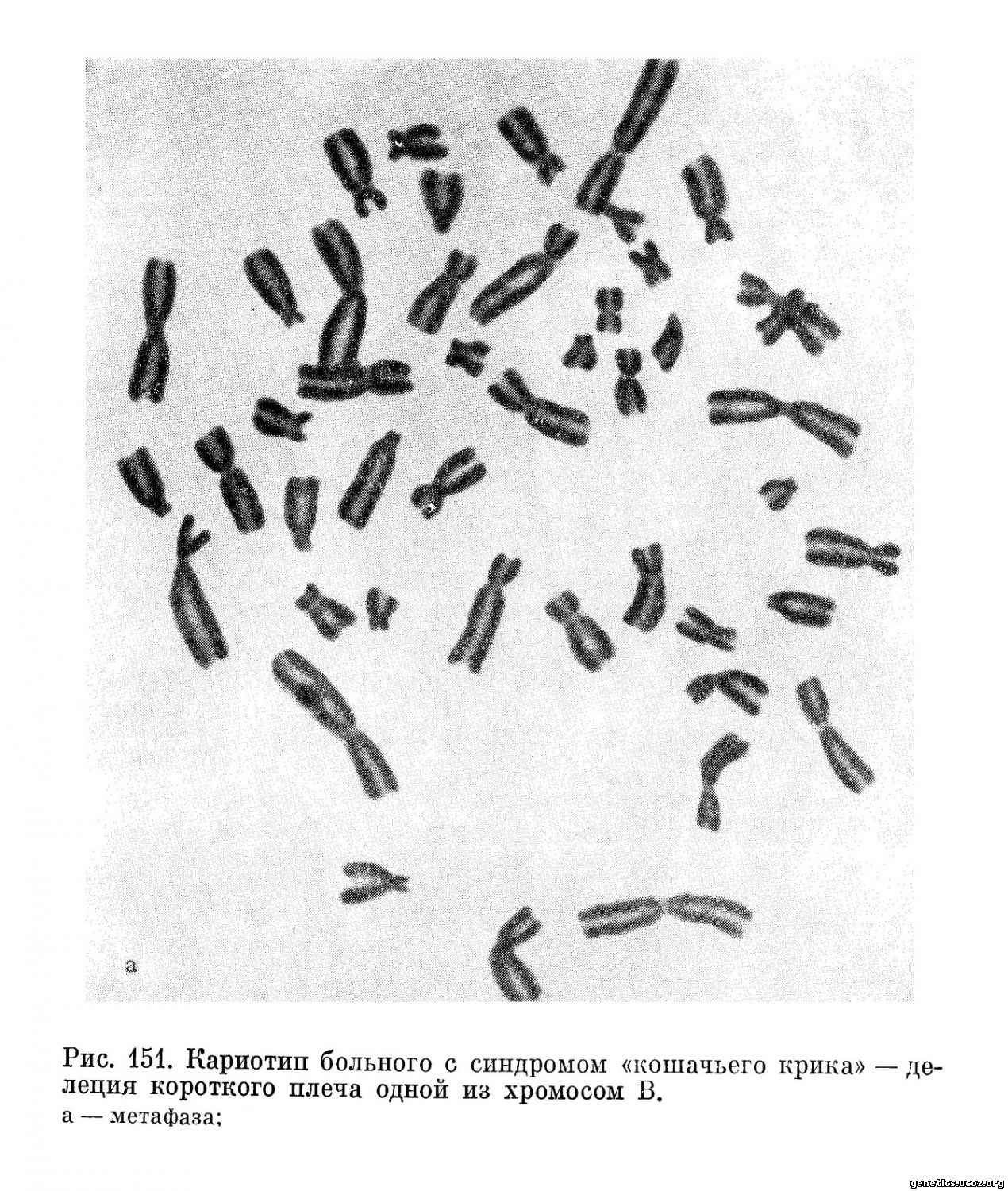
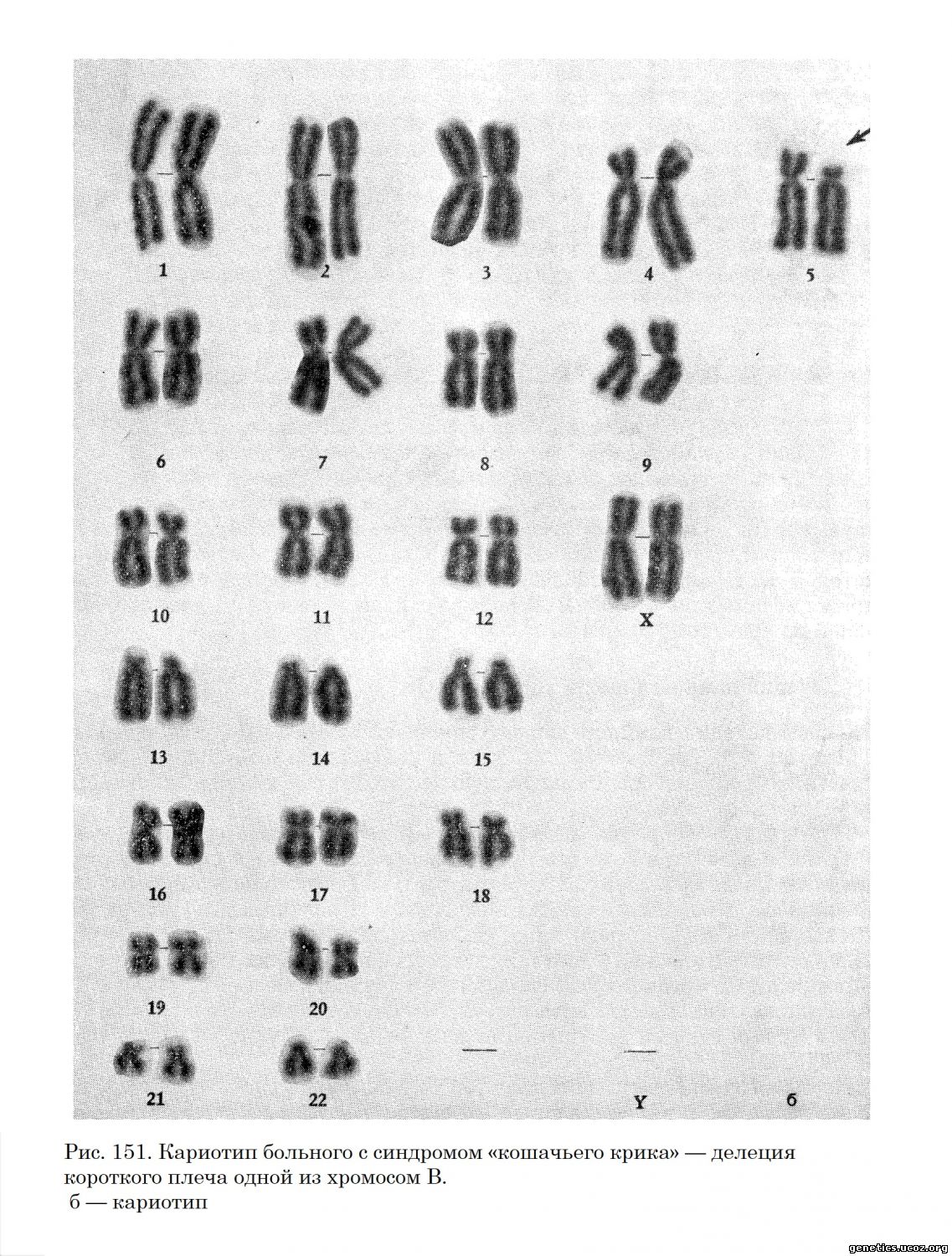
Не вызывающая сомнения общность клинической и цитогенетической картины позволила объединить все эти наблюдения в одну нозологическую единицу— синдром «кошачьего крика». Вскоре появились сообщения о симптоматике синдрома «кошачьего крика» у больных с несбалансированным кариотипом, родители которых имели сбалансированную транслокацию между хромосомами группы В и какими-либо другими (Reinwein, Wolf, 1965; Laurent, Robert, 1966), а также в случае кольцевой хромосомы из группы В. В некоторых семьях, имеющих такие транслокации, встречалось более чем по 1 больному.
Описаны и другие делеции в хромосомах этой группы. Wolf и др. (1965) наблюдали случай делеции короткого плеча одной из хромосом В, неотличимой цитологически от делеции, наблюдаемой в случае упоминаемого синдрома, однако симптоматика в данном случае не укладывалась в клиническую картину синдрома «кошачьего крика». По-видимому, произошла делеция короткого плеча хромосомы из другой пары, а не из той, делеция по которой дает синдром «кошачьего крика». К. Н. Гринберг с сотрудниками приводит описание 3 больных с подозреваемой делецией 1/5 длинного плеча одной из хромосом группы В.
б) Делеция длинного плеча хромосомы 18
Синдром делеции длинного плеча хромосомы из 18-й пары может служить примером того, как по мере накопления наблюдений из беспорядочного, на первый взгляд, набора симптомов вырисовывается клиническая картина определенного синдрома.
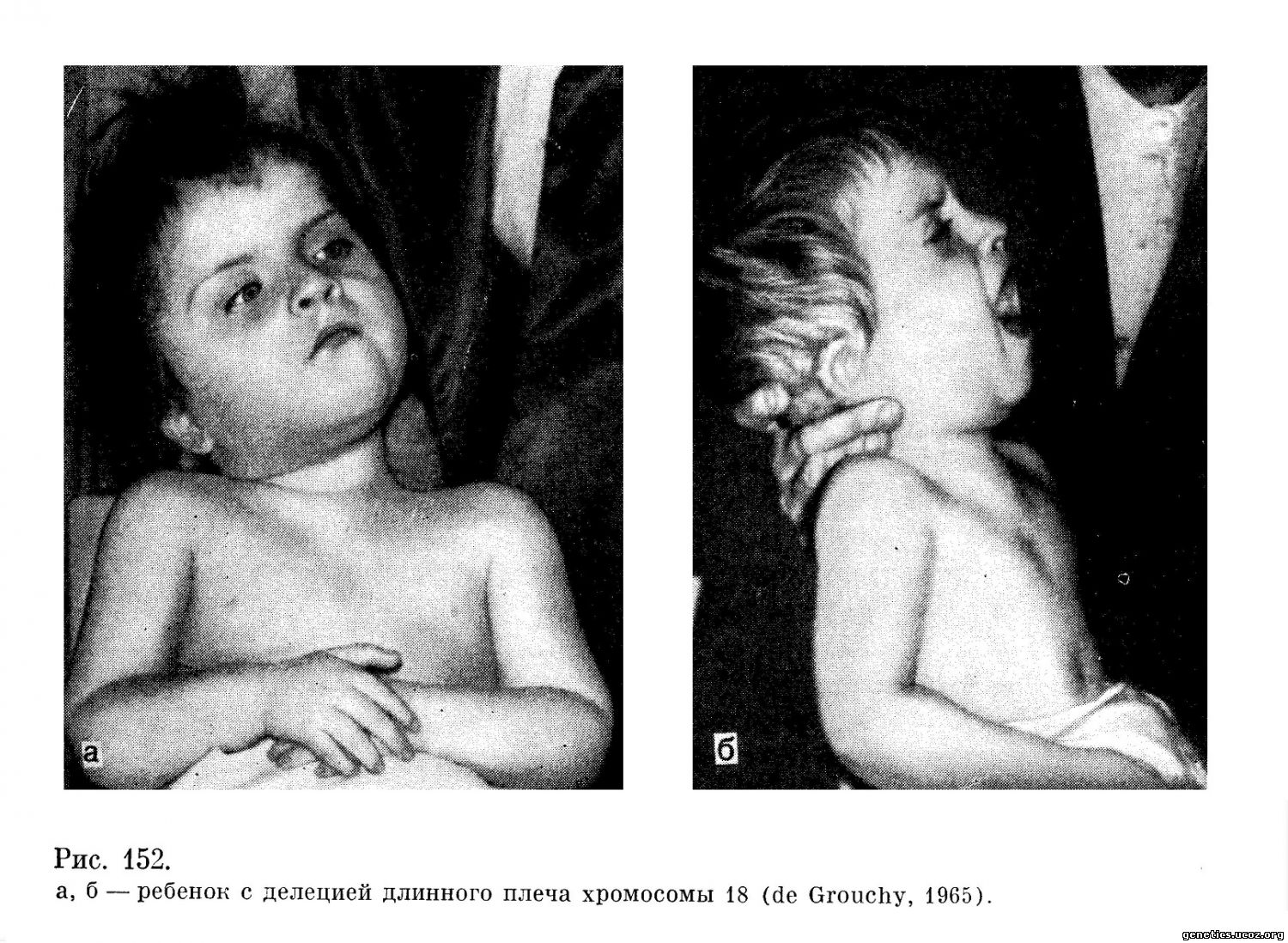
Первым эту аберрацию описал de Grouchy с соавторами в 1964 г. У больной девочки в возрасте 1 года с задержкой роста и умственного развития наблюдались следующие симптомы: легкая степень микроцефалии, арковидное небо, низко посаженные уши с выступающим антитрагусом, уменьшение средней трети лица, гипертелоризм. Кроме этого, у нее были обнаружены мышечная гипотония, гипотрофия, атрезия наружного слухового прохода и среднего уха, гипоплазия наружных половых органов. Имелись также аномалии развития почек, отеки ног и характерные для больных с хромосомными нарушениями изменения в рисунке кожных складок на ладонях.
Кариологическое исследование этой больной обнаружило у нее женский кариотип, полностью нормальный, за исключением того, что в группе Е не хватало одной хромосомы, по-видимому, Е-18, тогда как имелась одна хромосома — маленький субметацентрик, напоминающий по морфологии хромосомы группы F, но меньший по размеру (46, XX, Eq—). Цитологическая картина была интерпретирована как результат утраты части длинного плеча хромосомы из 18-й пары (рис. 152, 153).
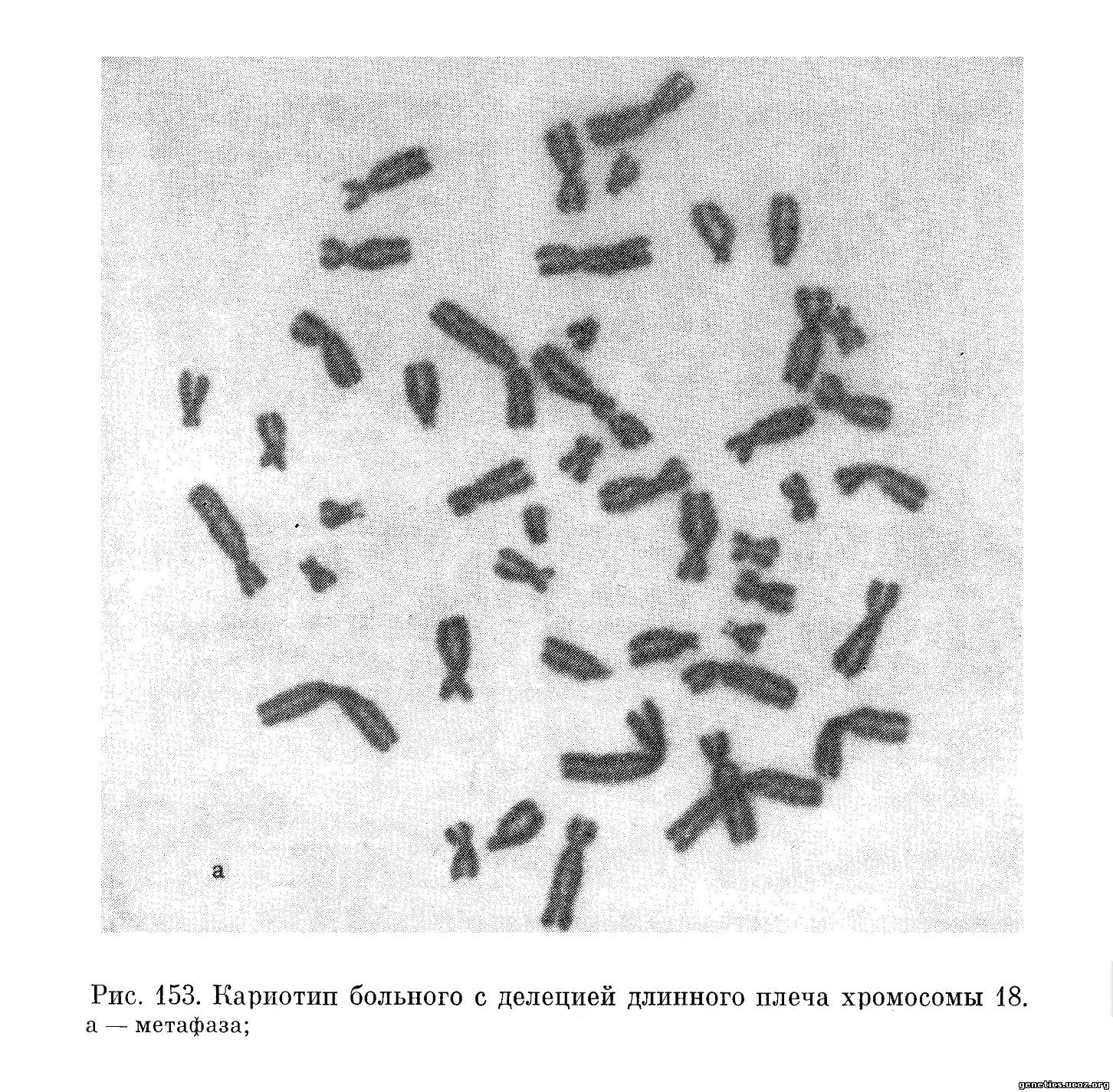

Это наблюдение оставалось единственным до 1966 г., когда были опубликованы данные обследования еще 7 больных (Wertelecki и др., 1966; Law, Masterson, 1966; Lejeune и др., 1966). Вместе с наблюдениями Insley, которые приводит Lejeune в сводке по этому вопросу, и с его собственными наблюдениями (Lejeune и др., 1967) в литературе имеется описание уже 12 больных (6 мальчиков и 6 девочек); из них у двоих имеется мозаичный по перестройке кариотип.
Сходство в клинической картине, как полагают Lejeune с сотрудниками, достаточно велико, а хромосомная аберрация полностью идентична у всех этих больных, что и позволяет выделить это заболевание в особую нозологическую единицу — синдром делеции длинного плеча хромосомы 18.
Основные симптомы этого заболевания приведены Lejeune. К ним относятся умственная отсталость, гипотрофия и гипотония, микроцефалия, характерные пропорции лица с сокращением его средней трети, низкий грубый голос, характерные аномалии ушей — уши низко посажены и имеют выступающий антитрагус. Гипертелоризм и эпикантус встречаются не обязательно. Достаточно часто, но не во всех случаях имеется гипоплазия наружных половых органов — у девочек отсутствие или недоразвитие малых губ и клитора, у мальчиков гипоспадия и крипторхизм. Примерно с такой же частотой (около половины описанных случаев) встречаются атрезия наружного слухового прохода, недоразвитие среднего уха, сердечные аномалии. Таким образом, это хромосомное заболевание можно считать вторым хорошо установленным синдромом, связанным с делецией части хромосомного материала, т. е. с частичной моносомией.
в) Делеция короткого плеча хромосомы 18
Делеция короткого плеча хромосомы из 18-й пары была впервые описана de Grouchy с соавторами в 1963 г. У 6-летнего мальчика имелась резкая умственная отсталость, его интеллект соответствовал таковому у ребенка 2—21/2 лет, однако он совершенно не мог говорить. У этого больного было круглое лицо с гипертелоризмом и эпикантом, расходящееся косоглазие, увеличенные уши, клинодактилия V пальцев рук и синдактилия III—IV пальцев ног. Рост больного и строение внутренних органов были нормальными. В результате кариологического анализа у него было обнаружено отсутствие одной хромосомы из пар 17—18, по всей вероятности из 18-й пары. Вместо этого присутствовала почти телоцентрическая хромосома размером с длинное плечо хромосомы 18 (46, XY, ? 18р—). В этой семье была очень сходная симптоматика у сестры больного, однако ее кариотип и кариотип их родителей оказались нормальными.
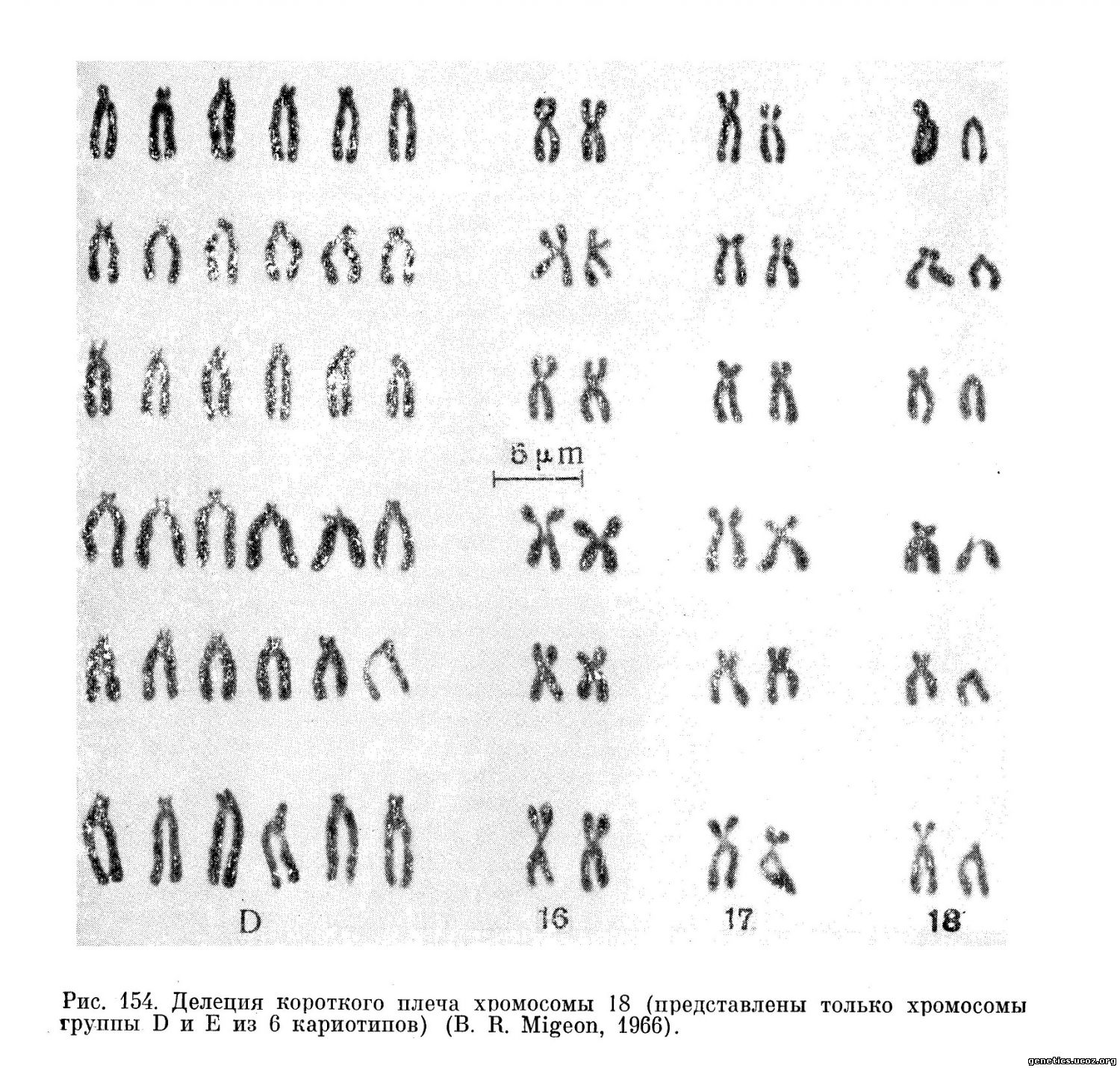
Еще одна делеция короткого плеча хромосомы 18 была описана Summit (1964). В этом случае у больной девочки в возрасте 4 лет наблюдалась симптоматика синдрома Тернера и до кариологического исследования ей ставился этот диагноз. У нее были обнаружены pterigium coli, круглое лицо, широкая спинка носа, гипоплазия супраорбитальной области, увеличенные уши, микрогнатия. Больная родилась от первой беременности у молодых родителей, у нее двое здоровых сибсов. Наркологически, как и в предыдущем случае, обнаружено отсутствие одной из хромосом 18-й пары и наличие телоцентрической хромосомы размером с длинное плечо хромосом 18-й пары (рис. 154) (46, XX, 18р—).
Наиболее интересный случай делеции короткого плеча хромосомы 18-й пары представлен в работе Uchida с сотрудниками (1965). Семья, описанная авторами этого сообщения, — первый и пока единственный пример унаследования делеции у человека. У матери 2 больных, имевших делецию короткого плеча хромосомы 18, был мозаичный кариотип (46, ХХ/46, ХХГ 18р—). В 88 из 100 проанализированных клеток у нее была обнаружена такая же делеция, как и у ее детей. К моменту обследования ей было около 33 лет и она родила 2 детей от внебрачных связей. Отцы детей разные. У одного ребенка обнаружены следующие симптомы: задержка умственного и физического развития, двусторонний птоз, эпикант, высокое небо, большие уши, кариес зубов, общая мышечная гипотония и др. У матери,, кроме умственной отсталости, имелась цебоцефалия и полная врожденная алопеция.
Migeon (1966) приводит еще один случай делеции короткого плеча хромосомы из 18-й пары. Этот случай интересен тем, что у больного, помимо указанных выше дефектов развития, имелась гепатоспленомегалия. Хотя в этом случае кариотипы родителей нормальны, особенности акушерского анамнеза матери (четыре беременности закончились выкидышами в первые 3 месяца беременности) наводят на мысль о возможном присутствии аберрантного клона клеток в гонадах одного из родителей.
В отличие от делеции короткого плеча хромосомы из группы В делеция короткого плеча хромосомы 18 не имеет четко очерченной клинической картины, достаточной для выделения ее в отдельную нозологическую единицу. По-видимому, это связано с недостаточным количеством наблюдений или с тем, что в некоторых случаях имеет место делеция короткого плеча хромосомы 17, а не 18, несмотря на то, что хромосомы этих пар идентифицируются более или менее уверенно.