Это заболевание является прекрасным примером тех превратностей, которые ожидают не одну хорошо разработанную проблему, и одновременно поучительным уроком, который преподает природа исследователю. Рассматривая историю исследований болезни Дауна, можно видеть, как рушатся, казалось бы, логичные концепции и оправдывается менее логичная, основанная на научной интуиции, точка зрения. Вместе с тем на примере этого заболевания можно видеть, как смещается эпицентр научных интересов в исследовании нозологической единицы благодаря одному, но фундаментальному открытию. Действительно, до 1959 г., когда французские исследователи и почти одновременно английские цитогенетики обнаружили хромосомную природу болезни Дауна, существовало так много теорий относительно ее происхождения, что это делало безнадежными дальнейшие попытки раскрыть ее природу, и лишь отдельные энтузиасты продолжали накапливать материал, касающийся этой патологии. Положение резко изменилось после открытия Lejeune и других авторов. Начиная с 1960 г. количество публикаций о болезни Дауна все возрастает.
Ввиду довольно значительной частоты этого заболевания, лучшей по сравнению с другими хромосомными синдромами выживаемостью, определенности клинической картины болезнь Дауна представляет огромный интерес для познания патогенетических механизмов хромосомных синдромов, для выяснения факторов, вызывающих нерасхождение хромосом, и для изучения ряда других проблем хромосомной патологии. Кроме того, именно вследствие высокой частоты этого заболевания представляется особенно важной научная разработка проблем, связанных с болезнью Дауна, с целью ее профилактики.
Впервые особую форму идиотии, сочетающуюся с аномалиями развития, описал французский психиатр Эскироль в 1838 г. (Turpin, Lejeune, 1965). В 1866 г. английский врач Лэнгдон Даун выделили это заболевание из других форм умственной отсталости как отдельную нозологическую единицу. Полное научное описание этой аномалии дали в 1876 г. англичане Фрейзер и Митчелл (Б. Лебедев, 1958). В дальнейшем болезнь Дауна описывалась в различных странах. В России об этом заболевании впервые сообщил в 1905 г. П. И. Ковалевский (цит. по Б. Лебедеву, 1958).
Как уже сообщалось, недостатка в теориях этиологии и патогенеза болезни Дауна не было. Как отмечает Б. Лебедев (1958), все коццепции относительно этиологии и патогенеза болезни Дауна можно подразделить на две группы: одни теории предполагают наличие дефекта в половых клетках, другие рассматривают эту аномалию как результат нарушения эмбрионального развития. Долгое время эти точки зрения считались взаимоисключающими, более того, концепция, придававшая большее значение наследственности, считалась наименее обоснованной. Болезнь Дауна рассматривали как типичный пример «бластофтории». Некоторые авторы (М. Я. Серейский, 1925) относили это заболевание к эндокринопатиям (плюригландулярная эндокринопатия). На основании эмпирического наблюдения того факта, что частота рождения больных увеличивается с возрастом матери и что больные, как правило, — последние дети в многодетных семьях, нашла распространение теория «изнашиваемости организма матери». Однако уже давно высказывались идеи, основанные больше на научной интуиции (базировавшейся в свою очередь на глубоких знаниях биологии и генетики), относительно того, что болезнь Дауна связана с аномалиями хромосомного аппарата. Приходится лишь удивляться прозорливости исследователей, высказавших такие суждения. В сущности цитогенетические механизмы болезни Дауна могли бы быть открыты еще в 30-х годах, так как «идеи носились в воздухе» и лишь несовершенство технических процедур мешало обнаружению прямых доказательств наличия хромосомных аномалий. Так, еще в 1932 г. Waardenburg предположил, что болезнь Дауна может быть вызвана дупликацией или нехваткой участка хромосомы. Віеуег (1934), проводя аналогию между трисомиками у Oenothera Lamarkiana, высказал предположение, что при болезни Дауна также может быть нарушено число хромосом и, как пишут Turpin и Lejeune (1965), «предоставив цитологам подтверждение этой гипотезы, указал, что возможное число хромосом при этой болезни — 49, 47, 50, 46».
Turpin и Caratzali (1937) уже прямо пишут о гипотезе «хромосомной аномалии», указывая в качестве примера возможного механизма на частичную трисомию по сегменту Ваг у дрозофилы гетерозиготных самок В/В+. Гипотезу хромосомной мутации выдвигал также Penrose в 1939 г.
Подтверждение в 1958—1959 гг. этих блестящих догадок принадлежит французским генетикам Lejeune, Gautier и Turpin (Институт прогенеза, Париж), которые обнаружили при этом заболевании 47 хромосом, причем лишней была хромосома из группы G. Это открытие сразу же перевело целый ряд концепций в разряд теорий, имеющих историческую ценность. В промежуток времени с июля 1958 г. по январь 1959 г. было обследовано около 10 больных и показано, что у них имеется 47 хромосом вместо 46.
Вслед за указанными авторами о подобных же находках сообщили в 1959 г. Jacobs с сотрудниками (6 случаев), а также Book с сотрудниками (3 случая). Эти открытия, о которых вскоре сообщили исследователи разных стран, не только открыли новую главу в патологии человека, но и пробудили интерес к старой проблеме болезни Дауна, заставив по-новому взглянуть на этиологию и патогенез этого заболевания и других «бластофторий».
а) Цитогенетика
Хромосомы группы G относятся к малым акроцентрикам. Эти хромосомы морфологически неразличимы в пределах своей группы. Так же как и хромосомы группы D, они являются спутничными, хотя спутники бывают выражены не на всех хромосомах и не в каждой метафазной пластинке (рис. 146).


Короткие плечи и спутники этих хромосом, по-видимому, целиком состоят из гетерохроматина (А. А. Прокофьева-Бельговская, 1966) и способны образовывать ассоциации как между собой, так и между хромосомами группы D и вторичными перетяжками и центромерными зонами других хромосом. Спутничные районы хромосом группы G, так же как и хромосом группы D, имеют отношение к ядрышку и называются «организаторами ядрышка». Согласно данным радиоавтографических исследований, эти хромосомы являются поздно метящимися. Хромосомы этой группы реже, чем все другие, повреждаются вирусами и химическими агентами (Nichols и др., 1964).
б) Частота заболевания
В отличие от других хромосомных синдромов болезнь Дауна встречается гораздо чаще, что позволило уже давно получить цифры частоты этой аномалии в популяции. Табл. 12, составленная по данным Penrose (1963), Turpin и Lejeune (1965), а также Е. Ф. Давиденковой (1966), дает представление о частоте этого заболевания по странам и у различных авторов.

Из приведенных в табл. 12 данных видно, что нет резких различий в частоте рождения больных с болезнью Дауна в разных географических и расовых популяциях.
Большинство авторов склоняется к той точке зрения, что частота болезни Дауна одинакова среди обоих полов. В подтверждение этого можно сослаться на наиболее обстоятельное сообщение Collman и Stoller (1961), которое охватывает 780 168 новорожденных. По данным этих авторов, среди 1134 больных соотношение полов было 105 : 100. Подобного же мнения придерживается Е. Ф. Давиденкова (1966) на основании анализа ленинградской популяции.
Если частота болезни Дауна распределяется равномерно по географическим зонам, расовым группам и полам, то этого нельзя сказать о распределении частоты этого заболевания во времени. Обширные исследования Collman и Stoller показали, что имеется периодическое варьирование частоты болезни Дауна, так что в отдельные периоды частота рождения больных большая, чем в другие (явления «Clustering»).
в) Возраст родителей
Уже давно было обращено внимание на корреляцию между увеличением возраста матери и рождением детей с болезнью Дауна. Shuttelworth (цит. по Turpin и Lejeuney 1965) еще в 1895 г. подметил, что почти половина больных появляется в многодетных семьях и рождается они последними. В 1905 г. этот автор показал, что матери больных детей были в среднем значительно старше матерей нормальных детей. Эти наблюдения привели к созданию теории «^изнашивания организма матери», которая допускала, что с возрастом и в результате многочисленных беременностей в организме женщины происходят изменения, вредно влияющие на формирование половых клеток, имплантацию яйца и развитие зародыша. Особенно значительный вклад в изучение проблемы связи возраста и частоты рождения детей с болезнью Дауна внес Penrose. Этот исследователь продемонстрировал чрезвычайно важный и интересный факт: частота рождения больных детей коррелирует с возрастом именно матери, а не отца. Ему удалось найти достаточное количество пар, где были пожилые отцы и молодые матери, а повышения частоты болезни Дауна среди их потомства не было (Penrose, 1963).
Приводимый график (Turpin, Lejeune, 1965), составленный по данным Penrose, хорошо демонстрирует зависимость частоты рождения больных детей от возраста матери (рис. 147 и табл. 13).
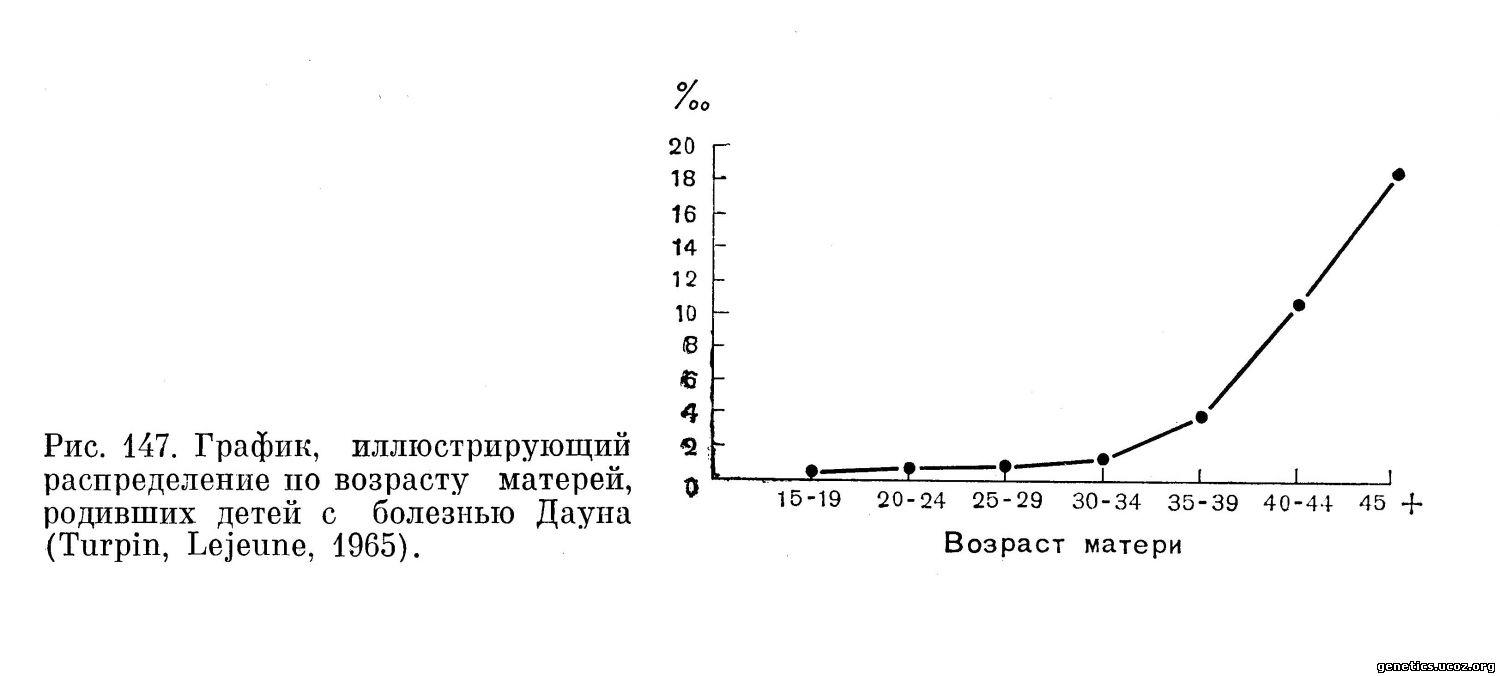
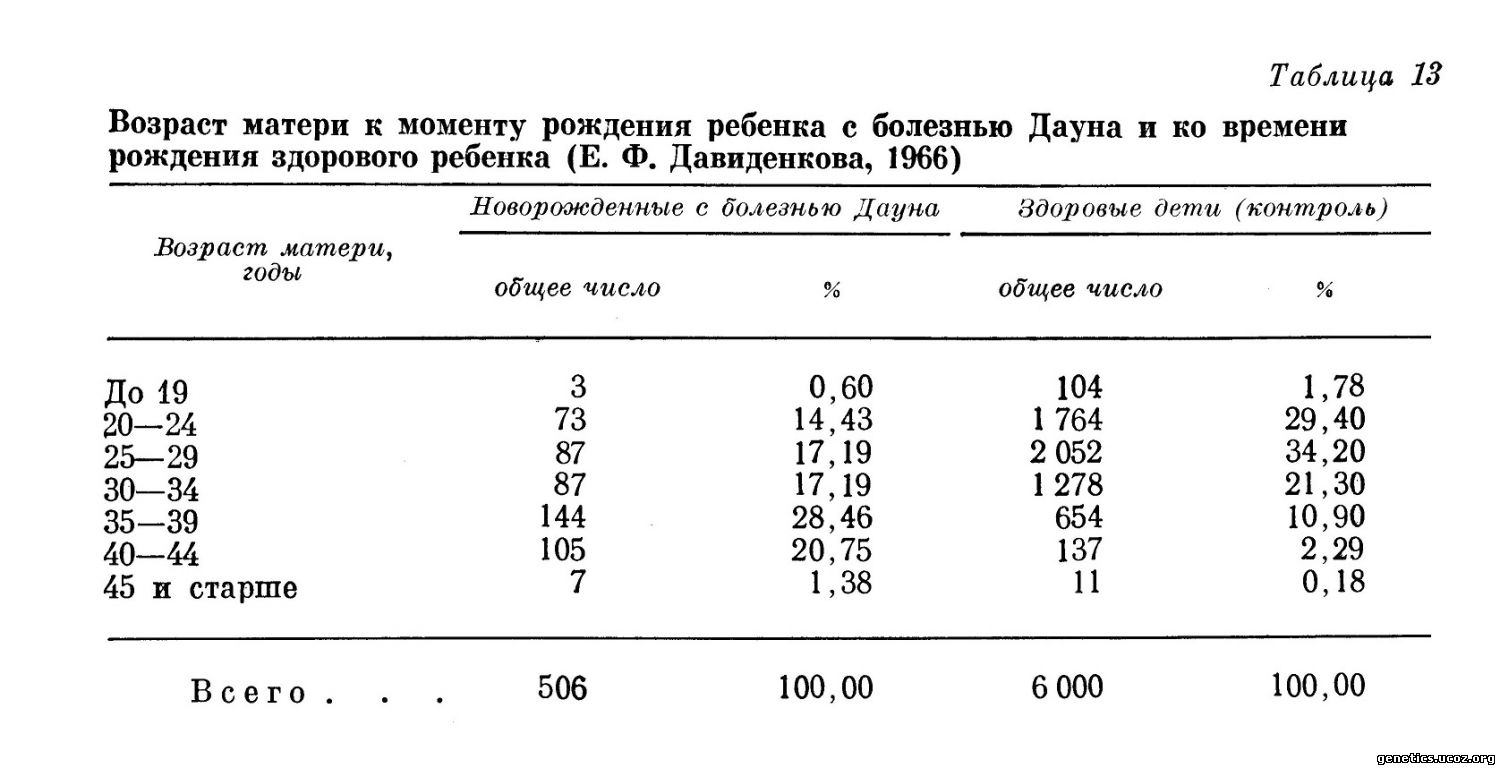
Важным обстоятельством является то, что больные дети рождаются и у молодых матерей. Еще Penrose в 1933 г. выделил группу молодых матерей, у которых дети страдали болезнью Дауна. В дальнейшем оказалось, что синдром Дауна вызывается не только классической трисомией, но и транслокацией одной из хромосом группы G на другую или на хромосому группы D. При такой транслокационной форме болезни Дауна нет корреляции с возрастом матери, как при регулярной трисомии.
г) Наследственность
Проблема наследственности при болезни Дауна может рассматриваться в разных аспектах. С преимущественно практическим аспектом этой проблемы приходится сталкиваться врачу-генетику, когда перед ним ставят вопрос о вероятности повторного рождения больного ребенка, если уже есть один, или в случае, когда есть один больной у близких родственников. Этот практический аспект побуждает исследовать вообще генетические механизмы нерасхождения. В этом разделе речь будет идти о собственно трисомии, тогда как транслокационная форма болезни Дауна, при которой имеется вполне определенная закономерность передачи по наследству транслокационной хромосомы.
д) Исследования близнецов
Сравнение конкордантности по какому-либо признаку однояйцевых (ОБ) и двуяйцевых (ДБ) близнецов часто и с успехом используется в медицинской генетике для проверки гипотез о роли наследственности в этиологии заболевания или о наследственной природе признака. В литературе опубликовано достаточно случаев, чтобы сопоставить конкордантность по болезни Дауна у ОБ и ДБ. Turpin и Lejeune обобщили близнецовый материал, опираясь в основном на обширные исследования Oster (1953), Allen и Baroff (1955), а также Carter и Evans (1961). Эти материалы представлены в табл. 14.

Высокая конкордантность по трисомии 21 у ОБ говорит о наличии анэуплоидной гаметы, которая после оплодотворения дала зиготу с трисомией, что обусловило трисомию, у обоих членов однояйцевой пары. Чрезвычайно важный факт для понимания механизма происхождения хромосомных аномалий представляет открытое французскими цитогенетиками (Turpin, Le- jeune, Lafourcade, Salmon, 1963; Wolff и др., 1963; Turpin, 1964) явление «гетерокариотипической монозиготности» (monozygotisme heterocaryoti- que). Указанные исследователи наблюдали несколько пар близнецов, которые были однояйцевыми по серологическим показателям и по трансплантационному тесту, но имели разный кариотип. В их числе описана пара близнецов, в которой один был нормальным мальчиком, а другой страдал болезнью Дауна и имеет трисомию хромосомы 21. При возникновении таких пар возможны следующие варианты: исходная зигота была нормальная, но во время первого деления дробления в одном бластомере, который дал начало одному близнецу, произошло митотическое нерасхождение; возможно также, что исходная зигота была с трисомией, но в первых же стадиях дробления произошла утеря лишней хромосомы. В результате такого «излечения» возник нормальный партнер. Такого рода наблюдения представляются важными потому, что демонстрируют возможность возникновения индивидуума, у которого все клетки имеют ненормальный набор хромосом, но не вследствие того, что этот индивидуум получил аномальный набор хромосом с одной из гамет, а в результате митотического нерасхождения в первом делении дробления. Фактически в данном случае мы имеем дело как бы с крайним случаем мозаицизма: если бы не произошло раннее разделение зиготы, то, вероятно, возник бы мозаичный индивидуум.
е) Потомство матерей с болезнью Дауна
В литературе описано больше 10 случаев беременности и родов у женщин с болезнью Дауна. Нередко эти беременности возникают в результате кровосмесительных актов. Turpin и Lejeune (1965) сообщают, что у 11 женщин с тписомией 21, описанных в литературе, было 13 беременностей, из которых 5 окончились рождением детей с трисомией 21. У 7 женщин родились нормальные дети, из них в одном случае — пара ОБ (Thuline, Priest, 1961), одна беременность закончилась рождением мацерированного плода и одна — рождением ненормальной девочки, однако без трисомии 21. Соотношение трисомиков и эусомиков в потомстве матерей-трисомиков (5:7) близко к ожидаемому соотношению 1: 1, так как матери-трисомики продуцируют два типа гамет — один тип содержит две хромосомы 21, другой — одну.
Следует отметить, что вторичное нерасхождение, по-видимому, не коррелирует с возрастом: матери-трисомики рождали детей-трисомиков в молодом возрасте, тогда как они сами были рождены пожилыми матерями (Carter, Evans, 1961).
ж) Семейные случаи
В разделе, посвященном Е-трисомии, сообщалось, что существуют наблюдения, касающиеся неслучайного сочетания в одной семье и у одного индивидуума нерасхождения по разным хромосомам. Наблюдения над семейными случаями болезни Дауна показали, что в большинстве таких случаев болезнь Дауна вызвана транслокацией типа G/G или D/G (Zellweger, 1962).
Priest с соавторами (1963) описал семью, в которой пробанд имел транс- локацию типа D/G, родители имели нормальный хромосомный набор, но тетка пробанда со стороны отца страдала болезнью Дауна, обусловленной стандартной трисомией 21.
Семейные случаи болезни Дауна приведены также в обстоятельных работах Penrose (1951), Hamerton с соавторами (1961), Carter и Evans (1961). Эти наблюдения навели на мысль, что у человека могут существовать гены, повышающие частоту нерасхождения, подобно тому как это было обнаружено у кукурузы (цит. по Kiossoglou и др., 1963) и у Drosophila melanogaster, у которой, так же как и у Dr. simulans, обнаружен ген claret-non-disjunction, который приводит к появлению в потомстве мух, имеющих этот ген, большого количества «исключительных» особей с нерасхождением. С целью обнаружения такого гена, который, по-видимому, должен быть рецессивным, Penrose (1962) предпринял большое исследование, которое охватило 600 с лишним случаев болезни Дауна. Если бы нерасхождение было обусловлено наличием рецессивного гена, то отмечалась бы большая частота близкородственных браков. Однако Penrose удалось установить только 3 случая близкородственной связи между предками отца и матери. В 5 случаях были обнаружены близкородственные браки среди предков отца и в 10 — среди предков матери. Интересно отметить, что пациенты с близкородственными браками в анамнезе матери родились в среднем от более молодых матерей, чем это обычно бывает при болезни Дауна. Этих данных совершенно недостаточно, чтобы сделать заключение о наличии рецессивного гена, приводящего к нерасхождению.
Следует отметить, что имеется еще несколько возможных механизмов, которые можно привлечь для объяснения случаев семейного накопления болезни Дауна (если исключить транслокационную форму). Наличие мелких структурных перестроек, не обнаруживаемых под микроскопом, может привести к нарушению конъюгации в мейозе и, следовательно, к нерасхождению. Подробнее этот механизм разбирается в разделе, посвященном структурным перестройкам.
Другой возможный механизм может быть связан с мозаицизмом, захватывающим гонады матери. В пользу такого механизма говорят факты обнаружения у родителей (преимущественно матерей) некоторых микросимптомов болезни Дауна (особенно это касается аномалий дерматоглифики). О подобных наблюдениях сообщали Turpin и Caratzali (1934), Turpin с соавторами (1947), Penrose (1964), И. Н. Штильбанс и Д. К. Берлинская (1966). Следует обратить внимание также на работы, непосредственно демонстрирующие наличие хромосомных аномалий, расцениваемых как проявление «семейной тенденции к нерасхождению», у матерей больных с трисомией 21 (Kiossoglou и др., 1963).
Таким образом, наследственная передача трисомии несомненна лишь при транслокационной форме болезни Дауна. Другие механизмы (наличие специальных генов, мелких структурных перестроек и роль мозаициз- ма родителей) нуждаются для обоснования в дополнительных фактах.
Касаясь роли наследственности при болезни Дауна, нельзя не упомянуть об интересном факте, сообщаемом Hirsch (1963). Показано, что имеется определенная корреляция между способностью ощущать вкус фенил- тиомочевины (ФТМ) и рождением детей с болезнью Дауна. Женщины, родившие таких детей, не ощущают вкуса ФТМ. Фенилтиомочевина и близкие к ней химические соединения оказывают специфическое антитиреоидное действие. Целый ряд авторов, в частности Fialkow (1964), полагает, что нарушения функции щитовидной железы способствуют нерасхождению хромосом. Наряду с этим известно, что вещества типа ФТМ содержатся в овощах. Поэтому возможно, что наличие генов, обусловливающих способность воспринимать вкус ФТМ, предохраняет от попадания в организм токсических продуктов (Hirsch, 1963), во всяком случае, чувствительность к ФТМ явно коррелирует со склонностью к определенному типу пищи (Glanville, Kaplan, 1965). В свете этих данных можно предполагать, что женщины, неспособные ощущать вкус ФТМ, потребляют пищевые продукты, содержащие антитиреоидные агенты, и гипофункция щитовидной железы приводит к появлению анэуплоидных гамет (возможно, в связи с ранним старением лиц, имеющих гипотиреоидизм).
з) Связь с заболеваниями матери и действием внешних факторов
В разделе, посвященном биологическим факторам как причине хромосомных аномалий, цитировались работы, доказывающие связь болезни Дауна с эндокринной патологией у матери. Имеются сообщения, которые, впрочем, не подтвердились, что заболевание матери вирусным гепатитом способствует рождению детей с болезнью Дауна. Мы имеем в виду работу Collman и Stoller (1962), которые наблюдали повышение частоты рождения больных после вспышек вирусного гепатита. Подобное явление наблюдали и другие авторы (см. раздел о Е-трисомии), однако связь с инфекцией не всегда устанавливалась. Относительно роли ионизирующей радиации четких данных нет. Некоторые авторы отмечают случаи диагностического облучения родителей незадолго до зачатия ребенка, но роль этого облучения не доказана. Schull и Neel (1962), изучавшие частоту болезни Дауна в Хиросиме и Нагасаки, не обнаружили повышения случаев рождения больных детей в облученной популяции.
и) Клинические особенности заболевания
Клиника болезни Дауна изучена достаточно хорошо и известна большинству врачей. Мы приводим краткое описание основных симптомов для того, чтобы можно было сравнить клинические особенности Gi-трисомии с другими хромосомными синдромами. При описании клинических особенностей этого заболевания мы основываемся на монографиях В. В. Русских (1963) и Е. Ф. Давиденковой (1966).
Беременность у женщин, родивших детей с болезнью Дауна, как правило, протекает нормально. Дети рождаются в срок и обычно имеют нормальные размеры тела и вес. Однако аномалии и дефекты физического строения столь характерны, что персонал родильных домов «узнает» этих больных сразу же при рождении.
Весьма характерны нарушения строения черепа. Голова ребенка уменьшена в размерах, череп круглый, затылок плоский. Брахицефалия и уплощение затылка увеличиваются с возрастом. Роднички закрываются поздно. Лицо уплощено, нос короткий с плоской переносицей и широким основанием. Верхняя челюсть недоразвита, что приводит к аномалиям прикуса. Особенно характерно строение глаз: глазные щели узкие, с косым разрезом. Имеется кожная складка — эпикант, идущая от верхнего века к нижнему во внутреннем углу глазной щели. Строение глазной щели напоминает то, что обычно наблюдают у представителей монголоидной расы, однако это сходство поверхностное. Отмечается узкое, высокое арковидное небо. Рот полуоткрыт, губы толстые, часто в трещинах. Характерен язык больных — он утолщен и покрыт глубокими поперечными бороздами. Часто отмечаются разнообразные аномалии зубов. Ненормальное строение голосовых связок приводит к изменению тембра голоса. Ушные раковины сформированы неправильно, отдельные части наружного уха бывают недоразвиты (рис. 148).

Часто наблюдаются аномалии в строении грудной клетки и скелета конечностей. Пальцы кисти и стопы укорочены, мизинец часто искривлен. Нередко отмечается синдактилия. Гипотония мышц и недоразвитие связочного аппарата приводят к возникновению характерного симптома — чрезмерной подвижности в суставах. При клиническом исследовании почти всегда обнаруживают порок сердца. Часто это выражается в дефекте межжелудочковой перегородки, тетраде Фалло или в незаращении боталлова протока. Примерно в половине случаев обнаруживается гипоплазия половых органов, однако к периоду полового созревания строение половых органов нормализуется. При эндокринологическом исследовании обнаруживается нарушение функции почти всех желез внутренней секреции.
Важнейшим симптомом болезни Дауна является умственная отсталость, выраженная в различной степени — от легкой дебильности до тяжелых форм идиотии. Дети с болезнью Дауна в ряде случаев могут обучаться во вспомогательной школе.
Эмоциональный фон больных своеобразен — они обычно добродушны, ласковы, легко отвлекаемы. М. Я. Серейский (1925) выделил два типа больных в зависимости от психического состояния — возбудимый и торпидный. Эти особенности он связывает с преимущественным нарушением функции надпочечников или щитовидной железы.
к) Патологическая анатомия
Патологическая анатомия этого заболевания подробно изучена В. В. Русских.
Специфических находок при этой патологии нет. Большинство исследователей отмечает аномалии и недоразвитие внутренних органов, особенно мозга. В соответствии с клинической картиной обнаруживают дефекты строения камер сердца и вторичные изменения, связанные с пороком сердца. Нарушения в строении элементов нервной системы позволили В. В. Русских сделать вывод о том, что болезнь Дауна представляет собой как бы антипод акромегалии.
л) Кровь и биохимические показатели
Turpin и Bernyer (1947) обнаружили, что у больных имеется аномалии сегментации ядер полинуклеаров. Как правило, это выражается в том, что ядра нейтрофильных лейкоцитов имеют меньше долей, чем в норме.
Valentine и Beck (1951) установили, что при хронической миелоидной лейкемии отмечается снижение активности щелочной фосфатазы в лейкоцитах. Так как при этой форме лейкоза имеется делеция части хромосомы 21, то естественно думать, что уменьшение активности фермента связано именно с делецией. Alter с сотрудниками (1963) обнаружил, что в лейкоцитах больных при болезни Дауна отмечается значительное повышение активности этого фермента. Отсюда был сделан вывод, что в хромосоме 21 расположен ген, ответственный за синтез этого фермента.
Brandt и др. (1963) установили, что при синдроме Дауна повышена активность фермента галактозо-1-фосфат-уридил-трансферазы. Это также дало основание для утверждений, что локус этого фермента находится именно в хромосоме 21.
Qershoff с соавторами (1958), Lejeune и др. (1960) обнаружили, что у больных с трисомией 21 имеется нарушение метаболизма триптофана, что выражается в пониженной по сравнению с контролем экскреции продуктов метаболизма триптофана (5-оксииндолуксусной, индолуксусной и ксантуреновой кислот). Эти наблюдения позволили сделать вывод, что у больных с синдромом Дауна нарушена активность оксикинуренин-трансаминазы.
Все эти данные позволяют думать, что гены, определяющие синтез соответствующих ферментов, локализованы в хромосоме 21. Однако против, такой точки зрения имеются возражения, которые нельзя не учитывать. Щелочная фосфатаза не является индивидуальным ферментом и правильнее говорить «щелочные фосфатазы». Генетическая регуляция этого фермента весьма сложна даже у Е. coli. Известно, что у человека и млекопитающих вообще уровень активности этого фермента зависит от влияния некоторых гормонов. Кроме того, активность фермента меняется по мере созревания клеток. Эти обстоятельства сильно мешают принятию соблазнительной, но слишком простой идеи о локализации структурного гена в хромосоме 21. Как уже указывалось, повышение уровня щелочной фосфатазы и галактозо-1-фосфат-уридил-транеферазы отмечается и при Еі-трисомии. Это обстоятельство еще более затрудняет принятие идеи о прямом соответствии между трисомией 21 и нарушениями активности этих ферментов.
м) Дерматоглифика
Дерматоглифика при болезни Дауна изучалась многими исследователями. Основные работы в этой области проведены Penrose и Lejeune. Следует отметить также вклад отечественных исследователей в эту проблему — Н. И. Штильбанс и Д. К. Берлинская собрали большой материал по дерматоглифике больных и их родственников.
Давая описания дерматоглифических признаков, мы опираемся в основном на работы Penrose (1963), Turpin и Lejeune (1965), Н. И. Штильбанса и Д. К. Берлинской (1966).
Для трисомии 21 характерно наличие глубокой поперечной складки на ладони («обезьянья складка», simian lines). Н. И. Штильбанс и Д. К. Берлинская считают более надежным диагностическим признаком наличие одной сгибательной складки на мизинце вместо двух в норме. Папиллярные узоры при болезни Дауна также изменены, вернее изменено соотношение различных типов папиллярных узоров. Однако эти признаки являются скорее качественными, их трудно измерить количественно. Более важным поэтому является положение трирадиуса — при болезни Дауна он расположен более дистально и поэтому имеет тупой угол (больше 57°). Дерматоглифический образец при болезни Дауна представлен на рис. 149).
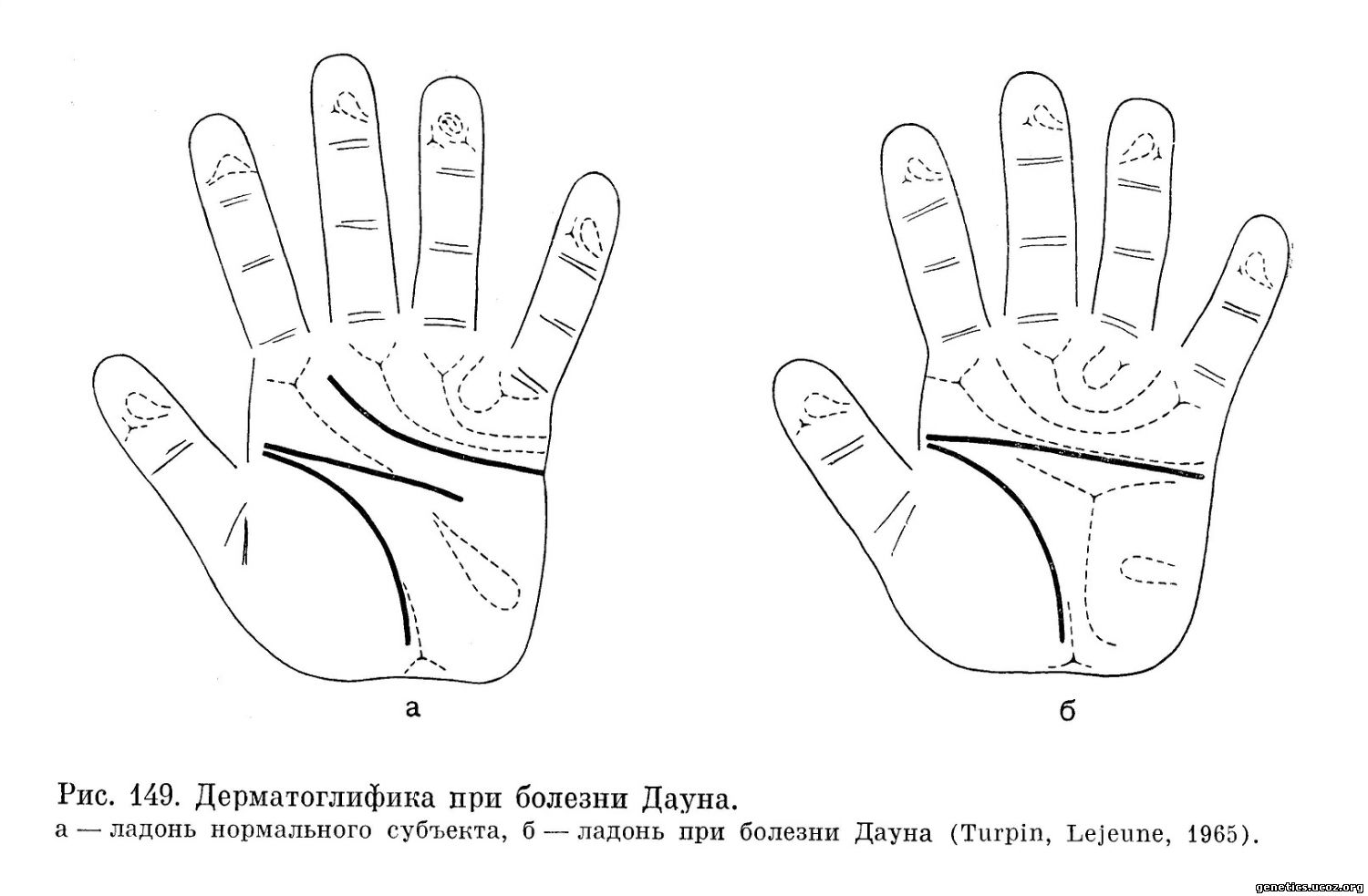
н) Диагноз, прогноз и лечение, медико-генетическая консультация
Диагноз болезни Дауна основан на характерном сочетании аномалий в физическом строении, данных цитогенетического и дерматоглифического исследования. Цитогенетическое исследование целесообразно проводить для исключения мозаицизма и обязательно у молодых матерей для обнаружения транслокации. Turpin и Lejeune (1965) считают, что случаев транслокаций среди всех случаев трисомий хромосомы 21 может быть от 1 до 2%.
Раньше больные с трисомией 21 погибали в детстве в связи с повышенной чувствительностью к инфекциям. Однако в настоящее время лица, страдающие этим заболеванием, доживают до 30 лет и более. Медико-педагогические меры и лечебные воздействия позволяют в какой-то степени корригировать имеющиеся дефекты и приучить больных к посильному труду. При лечении болезни Дауна применяются гормональные препараты (гормоны щитовидной железы и гипофиза), а также глютаминовая кислота (В. В. Русских, 1963; Е. Ф. Давиденкова, 1966).
При стандартной трисомии основными моментами, о которых должен помнить врач-генетик, являются средняя частота заболевания в популяции и различная частота рождения больных детей в разных возрастных группах матерей. По данным Е. Ф.
Давиденковой (Ленинград), риск для женщины родить ребенка с болезнью Дауна равен 0,11%. Для женщины 20—24 лет этот риск составляет 0,05% и в 18 раз больше для женщины в возрасте 40—44 лет. Однако практически следует разъяснить, что у женщины старше 40 лет риск рождения больного ребенка не превышает 1%. Чаще всего перед консультантом ставят вопрос о вероятности повторного рождения больного ребенка, если уже есть один больной. По данным различных авторов, в частности Carter и Evans (1961), риск повторного рождения больного ребенка выше у молодых матерей. Но даже при увеличении этого риска для молодых матерей вероятность практически не превышает 2%. Таким образом, при регулярной трисомии прогноз потомства благоприятен, однако исследование кариотипа обязательно, так как величина риска резко меняется при транслокации (см. соответствующий раздел) и при мозаицизме.
о) Мозаицизм
В 1961 г. Fitzgerald и Lycette (1961) описали мужчину 51 года, у которого отмечались некоторые, но не все признаки болезни Дауна. Почти половина исследованных клеток этого больного содержала 47 хромосом и лишней была хромосома из группы G; другая половина клеток имела нормальный мужской кариотип. В том же году Clarke с соавторами (1961) сообщили о длительном наблюдении над девочкой, которая развивалась психически нормально, но имела некоторые черты лица, свойственные болезни Дауна. При исследовании лейкоцитов периферической крови был обнаружен нормальный женский кариотип, но в клетках культуры кожи было 62% клеток с 47 хромосомами и лишней была хромосома 21. Науashi, Hsu и Chao (1962) описали мальчика с нормальным интеллектом и физическими признаками болезни Дауна, у которого в крови половина клеток содержала 47 хромосом с лишней хромосомой 21. Мать этого больного перенесла на III месяце беременности краснуху, однако это не может быть принято за этиологический фактор митотического нерасхождения, так как мозаичный клон наверняка возник раньше. Mauer и Noe (1964) сообщили о более сложном случае. У больного, которого они наблюдали, было три стволовых линии клеток — 46, 47 и 48 хромосом, причем соотношение клонов было 1:5:1. Лишними хромосомами были хромосомы группы G. Весьма интересны наблюдения Clark и сотрудников (1963) над судьбой клона с лишней хромосомой. У ребенка, обследованного ими в 1960 г., имелось 32% клеток с лишней хромосомой 21, в то время как через 3 года у этого же ребенка можно было обнаружить лишь 17% таких клеток.
До сих пор речь шла о мозаицизме, обнаруживаемом у более или менее ненормальных детей. Не менее важны и интересны случаи, когда мозаицизм обнаруживается у фенотипически здоровых родителей. Мы сошлемся на два сообщения такого рода. Blank с соавторами (1962) обнаружили мозаицизм у матери ребенка, страдающего болезнью Дауна. У матери явных симптомов заболевания не было. Smith и др. (1962) сообщили о мозаицизме у женщины, которая родила 2 детей, больных болезнью Дауна.
Из этих сообщений можно сделать некоторые выводы. По-видимому, существует значительное число больных (особенно лишь с некоторыми признаками заболевания), у которых имеется только часть клеток с хромосомной аномалией. Можно полагать также, что существует какая-то динамика пролиферации различных клонов, которая может привести к изменению состояния больного. Тот факт, что у некоторых мозаиков аномальные клетки обнаруживаются в коже и не обнаруживаются в крови, позволяет думать, что может быть и такой случай, когда мозаичный клон затрагивает ткани, недоступные цитогенетическому исследованию, и тогда возникает картина заболевания при нормальном кариотипе. В других случаях, очевидно, аномальный клон затрагивает гонады, и тогда такой индивидуум будет продуцировать анэуплоидные гаметы, что может явиться причиной «семейных» случаев.